 "
"
Team:Hawaii/Project/Part B MaterialsMethodsResults
From 2008.igem.org
(→Step 3: Conversion of pRL1383a into a Biobrick plasmid) |
(→Step 3: Conversion of pRL1383a into a Biobrick plasmid) |
||
| (8 intermediate revisions not shown) | |||
| Line 134: | Line 134: | ||
|}<br> | |}<br> | ||
In theory, transformants should be blue when plated on X-gal because of the α complementation of [http://partsregistry.org/Parts:BBa_J33207 J33207 (''lac promoter'' + ''lac Z'')] in DH5α. Transformation of DH5α with part [http://partsregistry.org/Parts:BBa_J33207 J33207] as provided in pSB1A2 yields blue colonies. However, none of the transformants obtained were blue, despite having the J33207 insert as confirmed by gel electrophoresis. The plasmid was sent for sequencing to verify the insert. | In theory, transformants should be blue when plated on X-gal because of the α complementation of [http://partsregistry.org/Parts:BBa_J33207 J33207 (''lac promoter'' + ''lac Z'')] in DH5α. Transformation of DH5α with part [http://partsregistry.org/Parts:BBa_J33207 J33207] as provided in pSB1A2 yields blue colonies. However, none of the transformants obtained were blue, despite having the J33207 insert as confirmed by gel electrophoresis. The plasmid was sent for sequencing to verify the insert. | ||
| - | + | [[Image:102908colonyPCRs.png|right|thumb|200px|EtBr stained 2% agarose gel ran at 95V for 1 hour. 3μl of transformed colony PCR product were loaded into each lane.]] | |
Sequencing of the converted plasmid by the Greenwood Molecular Biology Facility returned the following insert sequence: | Sequencing of the converted plasmid by the Greenwood Molecular Biology Facility returned the following insert sequence: | ||
:[[Image:BB1sequence.jpg|700px]] | :[[Image:BB1sequence.jpg|700px]] | ||
| - | + | <br> | |
There appeared to be a single base pair C->G transversion in the CAP binding site of the ''lac'' promoter. Otherwise, the sequence of the insert is identical to that of J33207. The ligation was repeated but no blue colonies were observed, yet again. Sequencing results of this new ligation is pending. | There appeared to be a single base pair C->G transversion in the CAP binding site of the ''lac'' promoter. Otherwise, the sequence of the insert is identical to that of J33207. The ligation was repeated but no blue colonies were observed, yet again. Sequencing results of this new ligation is pending. | ||
| - | To determine the ideal concentration of spectinomycin to use for isolation of single transformed colonies, an [[Team:Hawaii/Antibiotic_test_for_BB-pRL1383a|antibiotic test]] was carried out where equal amounts of transformed DH5α were plated on LB agar with varying spectinomycin concentrations. It was determined that sp<sub>40</sub> is the minimum concentration necessary for single colony isolation but sp<sub>100</sub> is the ideal concentration. At sp<sub>100</sub>, no untransformed colonies were observed and all colonies tested contained the correct insert. | + | To determine the ideal concentration of spectinomycin to use for isolation of single transformed colonies, an [[Team:Hawaii/Antibiotic_test_for_BB-pRL1383a|antibiotic test]] was carried out where equal amounts of transformed DH5α were plated on LB agar with varying spectinomycin concentrations. It was determined that sp<sub>40</sub> is the minimum concentration necessary for single colony isolation but sp<sub>100</sub> is the ideal concentration. At sp<sub>100</sub>, no untransformed colonies were observed and all colonies tested contained the correct insert.[[Image:102208plates.jpg|center|thumb|500px|K125000 transformed DH5α plated on LB supplemented with X-gal and various concentrations of spectinomycin.]] |
| - | + | ||
| - | + | ||
==Step 4: Device construction== | ==Step 4: Device construction== | ||
[[Image:Construct.jpg |right|thumb|200px|Figure 1: Construct of proposed GFP secretion device.]] | [[Image:Construct.jpg |right|thumb|200px|Figure 1: Construct of proposed GFP secretion device.]] | ||
The synthesized signal peptides and nirA promoter BioBricks were to be combined with at least three existing BioBricks to create two (or more) nitrate-regulated protein secretion devices according to the scheme detailed in Figure 1. The resulting devices would be placed in a ''Synechocystis'' compatible BioBrick vector derived from the RSF1010 derived plasmid pRL1383a. In the proposed devices, the signal peptides are be situated so they are in-frame with GFP. The translated polypeptide should consist of a N-terminal signal polypeptide leader sequence attached to a fluorescent protein. | The synthesized signal peptides and nirA promoter BioBricks were to be combined with at least three existing BioBricks to create two (or more) nitrate-regulated protein secretion devices according to the scheme detailed in Figure 1. The resulting devices would be placed in a ''Synechocystis'' compatible BioBrick vector derived from the RSF1010 derived plasmid pRL1383a. In the proposed devices, the signal peptides are be situated so they are in-frame with GFP. The translated polypeptide should consist of a N-terminal signal polypeptide leader sequence attached to a fluorescent protein. | ||
| - | [[Image: | + | [[Image:102508plasmiddigest.jpg|right|thumb|EtBr stained 4% agarose gel ran at 60V for 3.5 hours. Ten microliters of EcoRI/PstI digested DNA were loaded into each lane.]] |
Different degrees of success were experienced for ligation of parts. We were unsuccessful in ligating the ''pilA'' signal sequence to any BioBrick part. While we were able to ligate a transcriptional terminator to GFP fusion, this product was not successfully ligated to any other part. | Different degrees of success were experienced for ligation of parts. We were unsuccessful in ligating the ''pilA'' signal sequence to any BioBrick part. While we were able to ligate a transcriptional terminator to GFP fusion, this product was not successfully ligated to any other part. | ||
The parts that we were able to successfully ligate together are depicted on the right. All composite parts have been sequenced and confirmed. | The parts that we were able to successfully ligate together are depicted on the right. All composite parts have been sequenced and confirmed. | ||
| - | Of our intended constructs and controls, we were only able to create a ''lac'' | + | Of our intended constructs and controls, we were only able to create a constitutive secretion device under the control of the strong ''lac'' promoter. No transcriptional terminator was ligated to this construct because attempts at terminator ligation were unsuccessful. Instead, we relied on the terminators downstream of the insert on the plasmid. The sequence of the construct has been confirmed by sequencing by the Greenwood Molecular Biology Facility. Part functionality was demonstrated by green fluorescence observed in the transformed ''E. coli'' strain DH5α. |
| - | '''''lac'' promoter + rbs (efficiency 1.0) + ''slr2016'' + GFP fusion sequence''' | + | :'''''lac'' promoter + rbs (efficiency 1.0) + ''slr2016'' + GFP fusion sequence''' |
:[[Image:prsgsequence.jpg|left|700px]] [[Image:102808glowingprsgecoli.jpg|right|thumb|200px|''E. coli'' strain DH5α transformed with constitutive GFP expression device, in contrast to untransformed DH5α, as plated on LB+Amp<sub>100</sub>.]] | :[[Image:prsgsequence.jpg|left|700px]] [[Image:102808glowingprsgecoli.jpg|right|thumb|200px|''E. coli'' strain DH5α transformed with constitutive GFP expression device, in contrast to untransformed DH5α, as plated on LB+Amp<sub>100</sub>.]] | ||
We still plan on making the rest of the constructs and controls but recognize that they will not be completed in time for the Jamboree. | We still plan on making the rest of the constructs and controls but recognize that they will not be completed in time for the Jamboree. | ||
==Step 5: Testing for protein secretion== | ==Step 5: Testing for protein secretion== | ||
| - | The BioBrick vector can be inserted into ''Synechocystis'' sp. PCC6803 by triparental conjugation with ''E. coli'' harboring a transmissible plasmid (like RP1) and another ''E. coli'' containing our engineered plasmid. Plated ''Synechocystis'' sp. PCC6803 colonies successfully transformed | + | The BioBrick vector can be inserted into ''Synechocystis'' sp. PCC6803 by triparental conjugation with ''E. coli'' harboring a transmissible plasmid possessing a similar oriT (like RP1) and another ''E. coli'' containing our engineered plasmid. Plated ''Synechocystis'' sp. PCC6803 colonies successfully transformed will exhibit a glowing halo of secreted GFP. Transformed ''Synechocystis'' sp. PCC6803 grown in liquid media will result in fluorescent culture media. The efficacy of the signal peptides in transporting GFP into the extracellular media can be measured using a spectrofluorometer. |
We are in the process of inserting this construct into our modified pRL1383a plasmid, [http://www.partsregistry.org/Part:BBa_K125000 K125000], and transferring it into ''Synechocystis'' via conjugation. | We are in the process of inserting this construct into our modified pRL1383a plasmid, [http://www.partsregistry.org/Part:BBa_K125000 K125000], and transferring it into ''Synechocystis'' via conjugation. | ||
==Controls== | ==Controls== | ||
| - | A number of controls | + | A number of controls were to be constructed in parallel with our device to test device assembly and function. |
{| align="center" border="1" | {| align="center" border="1" | ||
!Construct | !Construct | ||
Latest revision as of 03:04, 30 October 2008
Step 1: Synthesis and assembly of the nirA promoter and pilA and slr2016 signal sequences
The nir promoter and the pilA and slr2016 secretion signal sequences were syntheized with the standard Biobrick sites. Oligonucleotide fragments of each were hybridized with its complement and ligated together to form whole, fully functional promoters and signal sequences. Assembly of these new Biobricks were verified by gel electrophoresis and sequencing.
nir promoter
| Oligonucleotide | Sequence | Length |
|---|---|---|
| pnir1_fb.syn.1 | CTAGAGCTAAATGCGTAAACTGCATATGCCTTCGCTGAGTGTAATTTACGTTACA | 55 |
| pnir2_fb.syn.1 | AATTTTAACGAAACGGGAACCCTATATTGATCTCTACTACTAGTAGCGGCCGCTGCA | 57 |
| pnir2_rb.syn.1 | GCGGCCGCTACTAGTAGTAGAGATCAATATAGGGTTCCCGTTTCGTTAAAATTTGTAAC | 59 |
| pnir1_rb.syn.1 | GTAAATTACACTCAGCGAAGGCATATGCAGTTTACGCATTTAGCT | 45 |
Signal sequences
| Oligonucleotide | Sequence | Length |
|---|---|---|
| pilA1_fp.syn.1 | CTAGATGGCTAGTAATTTTAAATTCAAACTCCTCTCTCAAC | 41 |
| pilA2_ff.syn.1 | TCTCCAAAAAACGGGCAGAAGGTGGTACTAGTAGCGGCCGCTGCA | 45 |
| pilA2_rf.syn.1 | GCGGCCGCTACTAGTACCACCTTCTGCCCGTTTTTTGGAGAGTTGAG | 47 |
| pilA1_rp.syn.1 | AGAGGAGTTTGAATTTAAAATTACTAGCCAT | 31 |
| slr2016-1_fp.syn.1 | CTAGATGGCAGCAAAACAACTATGGAAAATTTTCAATC | 38 |
| slr2016-2_ff.syn.1 | CTAGACCGATGAAGGGTGGAACTAGTAGCGGCCGCTGCA | 39 |
| slr2016-2_rf.syn.1 | GCGGCCGCTACTAGTTCCACCCTTCATCG | 29 |
| slr2016-1_rp.syn.1 | GTCTAGGATTGAAAATTTTCCATAGTTGTTTTGCTGCCAT | 40 |

Three different constructs of the full nir promoter and slr2016 and pilA signal sequences were created. The first followed ligation and restriction digest protocols using full concentrations of annealed products. The second was a ligation and restriction digest carried out using 10-1 dilutions of annealed product. It was suggested by Dr. Sean Callahan that if too much DNA was added to the ligation mixture, "DNA hairballs" would form between ssDNA (unannealed) and dsDNA, resulting in smears and multiple bands in the gel. The last method of construction simply placed equal concentrations of annealed products together. In theory, DNA overhangs would anneal appropriately, yielding an unligated but full promoter or signal sequence.
Ligation at full concentrations of annealed product worked best. The strongest bands were observed for this method. Multiple bands were still observed, above and below the expected band length (80-100bp) indicating both unligated DNA fragments and incompletely digested DNA or hairball structures. However, the strongest band for all three constructs corresponded to the desired product. The 10-1 ligations were barely visible on the gel, and multiple bands were still observed, so ligation and restriction digest efficiency was not improved by the addition of less DNA. The last method of annealing but not ligating yielded two bands in the gel corresponding to the two original DNA fragments.
The purified DNA was then ligated to a Biobrick vector, [http://www.partsregistry.org/Part:pSB1A2 pSB1A2], for subcloning in E. coli. Ligations were carried out using 3:1 molar concentrations of insert to vector.
Sequencing by the Greenwood Molecular Biology Facility returned the following results:
- nir promoter
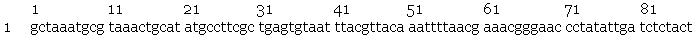
- slr2016 signal sequence

- pilA signal sequence
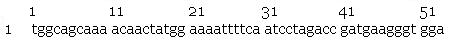
Step 2: Site-directed mutagenesis of GFP (BBa_E0040) into a fusion brick
GFP, as it currently exists in the Biobrick [http://www.partsregistry.org Registry of Parts], is a protein Biobrick, meaning that it will ligate out of frame with our signal sequence Biobricks. A primer was designed for site-directed mutagenesis of the GFP start codon to convert BBa_E0040 into a fusion Biobrick.

| Primer | Sequence | Length | G/C content | Tm |
|---|---|---|---|---|
| GFP (BBa_E0040) fusion / foward primer | GCCGCTTCTAGAcgtaaaggag | 22 bp | 54.55% | 60.2 C |
| GFP (BBa_E0040) fusion / reverse primer | cgagtcagtgagcgaggaag | 20 bp | 60% | 59.6 |
Following standard PCR protocol, [http://www.partsregistry.org/Part:BBa_E0040 GFP] was converted into the fusion brick, GFP fusion. GFP fusion was ligated into pSB1A2 and a colony PCR was conducted on a transformed colony. Following gel verification of the part, GFP fusion was sent out for sequencing.
Sequencing by the Greenwood Molecular Biology Facility returned the following sequence:

Step 3: Conversion of pRL1383a into a Biobrick plasmid
Part A of our project focuses on converting the RSF1010 based plasmid, pRL1383a into a sophisticated broad-host Biobrick plasmid. While we aim to ultimately express our secretion system in this new plasmid as part of a cyanobacterial expression system, we needed a workable shuttle vector between E. coli (where constructs will be made) and PCC6803 (the ultimate host). Converting pRL1383a into a much simpler Biobrick plasmid fulfilled this requirement. Verification regions, transcriptional terminators, and the Biobrick multiple cloning site (MCS) was be isolated from the [http://www.partsregistry.org/Part:pSB1A2 plasmid (pSB1A2)] containing BBa_J33207 via PCR. PCR primers also included HindIII and BamHI restriction sites for ligation into pRL1383a. This ligation replaced the original pRL1383a MCS which includes Biobrick and Biobrick compatible restriction sites. The MCS replacement was verified by restriction digest and plasmid sequencing.
| Primer | Sequence | Length | G/C content | Tm | Notes |
|---|---|---|---|---|---|
| HindIII-VF2BB | cctAAGCTTtgccacctgacgtctaagaa | 29 bp (20 bp) | 48.3% (50.0%) | 65.9 C (58.6 C) | Includes RE extension HindIII site and three 5' nucleotides for efficient cutting. Parentheses indicate primer information w/o RE site and 3 nucleic acids. Based on VF2 primer. |
| BamHI-VRBB | ccaGGATCCattaccgcctttgagtgagc | 29 bp (20 bp) | 55.2% (50.0%) | 67.9 C (58.0 C) | Includes RE extension BamHI site and three 5' nucleotides for efficient cutting. Parentheses indicate primer information w/o RE site and 3 nucleic acids. Based on VR primer. |
In theory, transformants should be blue when plated on X-gal because of the α complementation of [http://partsregistry.org/Parts:BBa_J33207 J33207 (lac promoter + lac Z)] in DH5α. Transformation of DH5α with part [http://partsregistry.org/Parts:BBa_J33207 J33207] as provided in pSB1A2 yields blue colonies. However, none of the transformants obtained were blue, despite having the J33207 insert as confirmed by gel electrophoresis. The plasmid was sent for sequencing to verify the insert.

Sequencing of the converted plasmid by the Greenwood Molecular Biology Facility returned the following insert sequence:

There appeared to be a single base pair C->G transversion in the CAP binding site of the lac promoter. Otherwise, the sequence of the insert is identical to that of J33207. The ligation was repeated but no blue colonies were observed, yet again. Sequencing results of this new ligation is pending.

Step 4: Device construction
The synthesized signal peptides and nirA promoter BioBricks were to be combined with at least three existing BioBricks to create two (or more) nitrate-regulated protein secretion devices according to the scheme detailed in Figure 1. The resulting devices would be placed in a Synechocystis compatible BioBrick vector derived from the RSF1010 derived plasmid pRL1383a. In the proposed devices, the signal peptides are be situated so they are in-frame with GFP. The translated polypeptide should consist of a N-terminal signal polypeptide leader sequence attached to a fluorescent protein.

Different degrees of success were experienced for ligation of parts. We were unsuccessful in ligating the pilA signal sequence to any BioBrick part. While we were able to ligate a transcriptional terminator to GFP fusion, this product was not successfully ligated to any other part. The parts that we were able to successfully ligate together are depicted on the right. All composite parts have been sequenced and confirmed. Of our intended constructs and controls, we were only able to create a constitutive secretion device under the control of the strong lac promoter. No transcriptional terminator was ligated to this construct because attempts at terminator ligation were unsuccessful. Instead, we relied on the terminators downstream of the insert on the plasmid. The sequence of the construct has been confirmed by sequencing by the Greenwood Molecular Biology Facility. Part functionality was demonstrated by green fluorescence observed in the transformed E. coli strain DH5α.
- lac promoter + rbs (efficiency 1.0) + slr2016 + GFP fusion sequence

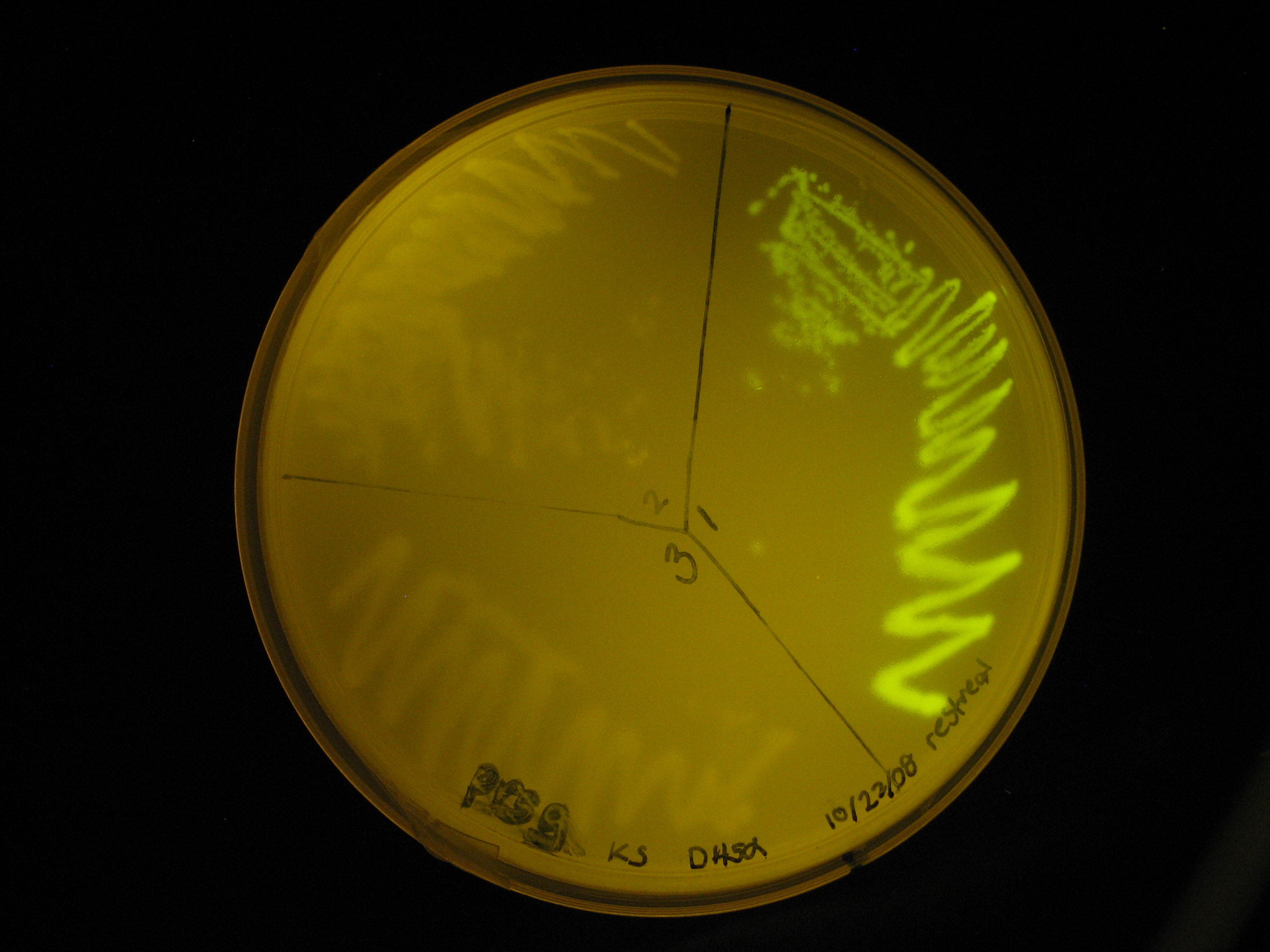 E. coli strain DH5α transformed with constitutive GFP expression device, in contrast to untransformed DH5α, as plated on LB+Amp100.
E. coli strain DH5α transformed with constitutive GFP expression device, in contrast to untransformed DH5α, as plated on LB+Amp100.


We still plan on making the rest of the constructs and controls but recognize that they will not be completed in time for the Jamboree.
Step 5: Testing for protein secretion
The BioBrick vector can be inserted into Synechocystis sp. PCC6803 by triparental conjugation with E. coli harboring a transmissible plasmid possessing a similar oriT (like RP1) and another E. coli containing our engineered plasmid. Plated Synechocystis sp. PCC6803 colonies successfully transformed will exhibit a glowing halo of secreted GFP. Transformed Synechocystis sp. PCC6803 grown in liquid media will result in fluorescent culture media. The efficacy of the signal peptides in transporting GFP into the extracellular media can be measured using a spectrofluorometer.
We are in the process of inserting this construct into our modified pRL1383a plasmid, [http://www.partsregistry.org/Part:BBa_K125000 K125000], and transferring it into Synechocystis via conjugation.
Controls
A number of controls were to be constructed in parallel with our device to test device assembly and function.
| Construct | Control | Test |
|---|---|---|
| nirA + [http://partsregistry.org/Part:BBa_B0030:Design rbs (BBa_B0034)] + GFP [http://partsregistry.org/Part:BBa_E0040:Design (BBa_E0040)] + txn term. [http://partsregistry.org/Part:BBa_B1006:Design (BBa_B1006)] | [http://partsregistry.org/Part:BBa_I14032:Design lac promoter (BBa_I14032)] + [http://partsregistry.org/Part:BBa_B0030:Design rbs (BBa_B0034)] + [http://partsregistry.org/Part:BBa_E0040:Design GFP (BBa_E0040)] + [http://partsregistry.org/Part:BBa_B1006:Design txn term. (BBa_B1006)] | Inducibility of nirA promoter |
| nirA promoter + [http://partsregistry.org/Part:BBa_B0034:Design rbs (BBa_B0034)] + pilA or slr2016 signal sequence + GFP fusion brick + txn term. [http://partsregistry.org/Part:BBa_B1006:Design txn term. (BBa_B1006)] | nirA or lac promoter + rbs [http://partsregistry.org/Part:BBa_B0034:Design (BBa_B0034)] + GFP [http://partsregistry.org/Part:BBa_E0040:Design (BBa_E0040)] + txn term. [http://partsregistry.org/Part:BBa_B1006:Design (BBa_B1006)] | Functionality of secretion peptides and GFP fusion |