 "
"
WetLab work
From 2008.igem.org
| HOME | Team | Project 1 | Project 2 | Parts | Modelling | Notebook | Doodle Board |
|---|
Contents |
CdLocalizer
Pathway
How CI and Cd sensor work?

Our construct

Experimental Procedure
1. Reconstruct pUC18 plasmid, insert GFP to the downstream of lac promoter
1.1 Use touchdown PCR to amplify GFP and add degradation tag and terminator.
Primers:
Forward: 5’- GA GAATTC G AGCAAGGGCGAGGAGCTGTTCACCG -3’
Reverse: 5’- CTTG CCCGGG TTATCACTTGTACAGCTCGTCCATGC -3’
(Template: provide by Guoqiang Chen’s lab in Tsinghua University)
1.2 Cut pUC18 plasmid with EcoR I and KpnI. Purify the cut pUC18 from agarose gel with Kit.
1.3 Purify the PCR product GFP from agarose gel with Kit and cut with EcoR I and Kpn I.
1.4 Purify the cut GFP with column.
1.5 Run a gel to compare the concentrations of the fragment and vector in order to decide their volume in the ligation mixture.
1.6 Incubate the ligation mixture at 16 degree.
1.7 Use 10μl ligation mixture to transform 200μl DH5αcompetent cell, spread on the LB plate(Amp), and incubate at 37 degree over night.
1.8 Pick three clone to 5ml LB and shake at 37 degree for 12h, miniprep the reconstruct plasmid.
1.9 We sequenced the inserted part and it was right.
2. Synthesize the Nde I-Pm(complementary chain)-Pst I-Xba I-cad-Spe I-RBS(strong)-CI+LVA tag-Sac II-T1, T2-BamH I (the Blue part is the synthesis sequence) sequence. We use synthesis because we cannot use PCR to get the sequence from Staphyloccocus aureus Rosenbach
3. Insert CheZ(with tag and terminator) into the reconstruct pUC18(with GFP)
3.1 Extract the genome of E.coli DH5αstrain and use PCR amplify CheZ from it.
Forward primer:
5’-GTCATGCC CA TATG CAACCATCAATCAAACCTGC-3’
Reverse primer:
5’-AT AGG CCT AAATCCAAGACTATCCAACAAATCGTCCACCTGATC-3’
3.2 Purify the PCR product from gel and then cut with Nde I
3.3 Cut the pAcYc duet-1 plasmid with EcoR V and Nde I
3.4 Run a gel to compare the concentration of the vector and insertion in order to decide the their volumes in the ligation mixture
3.5 Ligate the vector and the cheZ, transformate DH5α and spread the LB plate(Chl).
3.6 Pick three clones to 5ml LB(Chl) and shake at 37 for 12h, then miniprep the reconstruct the plasmid.
3.7 Synthesize the Stu I-tag-Hind III-T0/T1T2-AatII sequence (with stick end), which contains CheZ’s tag and terminator. We firstly synthesized the sequence as two single strings, and then annealing them together as a double string DNA.
The two single strings’ sequence is below:
Forward
5’-CCT GCTGCAAACGACGAAAACTACGCTTTAGTAGCT TAA A AGCTT
Stu I end LVA tag stop codon Hind III
CGCAAAAAACCCCGCTTCGGCGGGGTTTTTTCGC GACGT-3’
T0 Aat II end
Reverse
5’- C GCGAAAAAACCCCGCCGAAGCGGGGTTTTTTGCG A AGCTT TTA
Aat II end T0 Hind III stop codon
(complementary)
AGCTACTAAAGCGTAGTTTTCGTCGTTTGCAGC AGG
LVA tag (complementary) Stu I end
3.8 Cut the pAcYcduet-1-cheZ plasmid with StuI and Aat II and column purify the vector
3.9 Run a gel to compare the concentration of the vector(pAcYcduet-1-cheZ) and insertion (Stu I-tag-Hind III-T0/T1T2-AatII) to make sure their volumes in the ligation mixture.
3.10 Ligate the two parts transformation spread plate pick 4 clones shake and get the reconstruct plasmid
3.11 We sequenced it and it was right.
3.12 Then we cut the whole CheZ-tag-terminator with NdeI and AatII and ligate the fragment into the pUC18-GFP vector, which is cut by the same two enzymes.
3.13 This sequenced this plasmid and the sequence was right.
4. Cut the synthesized Nde I-Pm(complementary chain)-Pst I-Xba I-cad-Spe I-RBS(strong)-CI+LVA tag-Sac II-T1, T2-BamH I (the Blue part is the synthesis sequence) sequence with BamH I and Nde I, the same was done on the vector(pUC18-cheZ-tag-ter-GFP). Then ligate them together and got a new reconstruct plasmid. It was also sequenced.
5. Put RBS-CI-tag into last vector.
5.1 Use PCR to amplify CI from lambda DNA, adding RBS to 5’ and tag to 3’
First round primers:
5’-GAGGGGACAAactagt ATGAGCACAAAAAAGAAACCATTAACACAAGAGC-3’
5’-TCGTTTGCTGCAGGCCT gccaaacgtctcttcaggccactgac-3’
Second round primers:
5’-C A CTAGT tctagaGAAAGAGGGGACAAactagt ATGAGCACAAAAAAGAAACC-3’
5’-ACT CCGC GG TTA AGCTGCTAAAGCGTAGTTTTCGTCGTTTGCTGCAGGCCT gccaaacgtctcttcagg
We also did a point mutation with PCR to mutate a Hind III restriction cite.
Forwad:
5’-GGCTCCAAGCCTAGCTTTCCTGACGGAATG-3’
Reverve:
5’- cattccgtcaggaaagctaggcttggagcc-3’
5.2 Cut the RBS-CI-tag and the reconstructed vector with Sac II and Spe I and ligate them together.
5.3 Transformation the final plasmid to the ΔCheZ E.coli stain.
Now we have the reconstructed bacteria!!!
DualReceptor
1. Project objects
The aim of this project is to guide E.coli to a certain concentration of xylene/toluene, not too high or too low, so that the recombined E.coli can not only indicate the xylene/toluene pollution but also can survive in a not lethal concentration of pollution.
2. Pathway in the recombined E.coli

Figure1. Pathway in the recombined E.coli When there is xylene/toluene in the environment, the xylR receptor binds to the xylene/toluene molecular and becomes a active transcription factor, which activate promoter Pu. As a result, cheZ and xylCMAB genes are transcripted and translated. The cheZ protein can make the E.coli to move to higher concentration of xylene/toluene.
When the xylCMAB are activated, the several enzymes which degenerate toluene to benzoate are expressed. As a result, the benzoate can activate receptor xylS and activate promoter Pm and therefore induce the reverse transcription and an antisense mRNA is formed. The antisense mRNA will quickly block the original mRNA of cheZ, and therefore ceases the expression of cheZ protein. Then the E.coli would stay at the same place. The xylR, xylS, xylC, xylM, xylA, xylB gene and promoter Pu, Pm all come from Pseudomonas putida mt-2 plasmid pWW0. The original gene patterns are shown in the following figure.
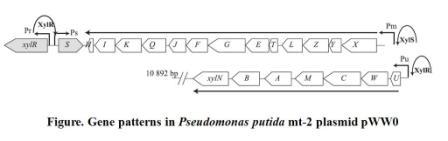

Figure2. Gene patterns in Pseudomonas putida mt-2 plasmid pWW0
3. Construct
xylR and xylS will be constructed into pACYCDuet-1(Cm, p15A ori) Pu-xylCMAB and Pu-cheZ-Pm will be constructed into pUC18(Amp, pMB1 ori) Several couples of primers has been designed. Pu and Pm are nearly 200bp long each, and we use PCR to ligate them to the Pu-xylCMAB and Pu-cheZ-Pm.
4. Knockout of cheZ

One single colony has its cheZ knocked out, replacing by kana gene. (Line 3&7) Another colony is a wide type, and has a 700bp cheZ gene.(Line2&6) RP1616 is from a foreign lab(line1). Its 700bp cheZ gene has been replaced by a 300bp segment.
TheoRiboSwitch
Recombinant Plasmids

CheZ is introduced into this plasmid under the control of riboswitch.To help this operon to be transcripted in non-BL21 E.coli strains, the gene coding for prokaryotic T7 RNA polymerase under the control of an artificial promotor is also introduced into this plasmid.

LacI with an LVA tag sequence is introduced into this plasmid.
Molecular Cloning Experiment
The whole pathway is designed as follows [Fig.3]:
Figure 3. The designed chemotaxis pathway.
When theophylline come, the translation is activated, the cheZ is expressed, so the bacteria can move forward. However, without theophylline, cheZ is blocked, so the bacteria can only tumble. Taken these together, the bacteria will behave as chemotaxis to the theophylline, which is our goal. In addition, to mimic the the bacteria’s natural ability of adaptation to the environment, we introduce a feedback mechanism: A lacI gene is also controlled by the TARS, when it is activated, it will block cheZ’s transcription.
The strategy of the pathway construction is mainly use PCR to combine the all elements together. Take the TARS-cheZ part for example [Fig. 4]
Figure.4 The procedure of construction. Form left to right, each blocking represents as T7 promoter, lacI binding site, TARS, cheZ, LVA degradation tag, T7 terminator, respectively. The arrows above are primers.
First step, we use TARS-plasmid as template, use primer Z01 & Z04, and add the lacI binding site to the upstream of TARS. Then we use the product of the 1st round PCR as template, and Z02 & Z04 as primers, to add the T7 promoter to the 5’ region of the fragment. By the same way, we use pET-28 plasmid as template of lacI, and add the LVA degradation tag and T7 terminator to the 3’ region in successive two round PCR, with the primers Z05 & Z06 and Z05 & Z07, respectively. After each PCR, we use gel electrophoresis to check the products, and use Gel Extraction Kit to purify them.
Secondly, we conduct another PCR. Since the primer Z04 has an overlapping with Z05, so the TARS product’s 3’ region can hybridize to the 5’ region of lacI product. So at first 10 cycles of the reaction, we add two products as template without primers, the two fragments will fuse to a longer one. After that, we add primers Z02 & Z07, the longer fragment will be proliferated, these are the final product we want.
With the same methods, we design another set of primers, to construct the second device. The components of the second device is almost the same as the first one, except that the lacI gene is replaced by the cheZ gene and T7 terminator is not added. The cheZ is cloned from the genome of E.coli K12 strain.
As we designed, the EcorI site is put into 5’ end of TARS-lacI part, and SpelI into 3’ end. BglII and Bpu1102I sites are put into 5’ end and 3’end of TARS-cheZ part respectively. After restriction enzymatic digestion, the TARS-lacI part is inserted into the PACYC177 vector, and TARS-cheZ part is inserted into pET-15b(lacI-) vector. In order to make our system work in a strain that does not have a T7 polymerase, we also add a T7 polymerase gene into the TARS-cheZ-pET-15b, restriction enzyme sites of this part are EcorI and HindIII. The promoter that control the T7 polymerase expression comes from the Part J23108. [Fig.5]
Plate check
Procedure
Swarming assay
Experimental Procedure
1. Prepare the competent cells of different strains of E. Coli, e.g.BW25113, BW25113 (CheZ), Rp1616, BL21 etc. using standard method.(Calcium Chloride)
2. Tranform the competent cell with plasmids previously contructed,e.g. pET15b-TARS-CheZ, pET15b-CheZ-T7, PACYC177-TARS-lacI etc.
3. Apply 2uL bacteria medium to a 0.4% agar plate (60mm dish) in the presence or absence of Theophylline. The final concentration of the theophylline should be around 3~10mM. IPTG is also added to the system, to afinal concentration of 5mM.
4. Incubate the plates in a 30 degrees C incubator for 10~24hrs and take photographs.
5. Compare the radius of the swarming plaque of each stain to test the efficacy of the molecular device transformed.
Additional attractant gradient plates can also be used.
Results
To be expected.
| HOME | Team | Project 1 | Project 2 | Parts | Modelling | Notebook | Doodle Board |
|---|