Culture of Stable strain with biobricks 2008
- Streaks on plates with LB and the adapted antibiotics to isolate colonies
- Incubate O/N at 37°C
- Take clone with a toothpick and put in 7.5ml LB with adaptated antibiotics (LB have to be prepared before !!!)
- Will be use for Miniprep and Stock in glycerol
- 2-3 clones isolated by Biobricks
- Incubate O/N at 37°C (open just a little the cap for bacteria's breathing)
- Don't store LB in the fridge even with antibiotics !
Glycerol Stocks
- Remove 2.5mL of each culture and centrifuge.
- Discard the supernatant and resuspend pelleted bacteria in 1mL of LB.
- Add 500µL of 60% glycerol.
- Store at -20°C.
Minipreps (QIAGEN kit)
- centrifuge 5mL of culture 8 min at 3,500 to 4,000 g.
- Resuspend pelleted bacterial cells in 250 µl Buffer P1 and transfer to a microcentrifuge tube.
- Add 250 µl Buffer P2 and mix thoroughly by inverting the tube 4–6 times. If using LyseBlue reagent, solution turns blue.
- Add 350 µl Buffer N3 and mix immediately and thoroughly by inverting the tube 4–6 times. If using LyseBlue reagent, solution turns colorless.
- Centrifuge for 10 min at 13,000 rpm (~17,900 x g) in a table-top microcentrifuge.
- Apply the supernatant (from step 4) to the QIAprep spin column by decanting or pipetting.
- Centrifuge for 30–60 s. Discard the flow-through.
- Recommended: Wash the QIAprep spin column by adding 0.5 ml Buffer PB and centrifuging for 30–60 s. Discard the flow-through. This step is only required when using endA+ or other bacteria strains with high nuclease activity or carbohydrate content (see QIAprep Miniprep Handbookfor more details)
- Wash QIAprep spin column by adding 0.75 ml Buffer PE and centrifuging for 30–60 s.
- Discard the flow-through, and centrifuge for an additional 1 min to remove residual wash buffer.
- To elute DNA, place the QIAprep column in a clean 1.5 ml microcentrifuge tube. Add 30 µl Buffer EB or water to the center of each QIAprep spin column, let stand for 1 min, and centrifuge for 1 min.
Electrophoresis
An electrophoresis can be done to check if there is Product of Miniprep
- Gel : 1% agarose with BET added (5 µL BET for 100 mL TBE)
- 10 µL Quick-Load 1 kb DNA Ladder
- 2 µL LB + 3 µL DNA
Concentration of the Miniprep
By biophotometry
- Blank : 55 µL of pure water + 5 µL EB
- Sample : 55 µL of pure water + 5 µL of DNA
Check if the ratio 260/280 is over 1,6
Think about the dilution !
Digestion
- 1 µg of plasmid / 250 ng of PCR product
- Buffer (n°2) 10X : 3µL
- BSA 100X : 0.3µL
- Pure water qsp 30 µL
- 1 µL of each enzyme
- Incubate during about 2h30-3h at 37°C, then 20 minutes at 65°C (to inactivate the enzymes).
Migration after digestion
- Run the whole samples (30 µL) in a 1-2% agarose gel with TAE buffer (a new one)
- Run at 50 V until halfway
- 10 µL of ladder 1kb and 100 pb on every side
- 30 µL of DNA + 6 µL of LB
Separate each band by an empty one !
- For each new extraction it's important to have a new bath of TAE Buffer for the run and of ETB for the revelation
- Use UV at lower intensity and for shorter time as possible
- Use a new blade for each extraction
- The band weight must be less than 400 mg
Amplification of promoters
(to amplify the sequence in order to have enough amount of DNA to carry out the following of our experiments)
- Preparation of the templates : Resuspend of 1 colony in 100µl of water.
- Preparation of PCR mix :
For each samples
1 µl dNTP
10 µl Buffer Phusion 5x
2,5 µl Oligo_F
2,5 µl Oligo_R
1µl template
1 µl Phusion
50 µl qsp H2O (33µl)
- make a mix with buffer, oligos and water for n+1 samples
- negative control : without template
- positive control : known template
LID : 105°C
1. 98°C 30 sec initial denaturation
2. 98°C 10 sec denaturation
3. 60°C (depending of the size of oligos) 30 sec annealing
4. 72°C (1 min for 1 kb)
5. go to : 2 rep : 24-29
6. 72°C 5 min
7. sound : 1
8. hold : 10°C
Purification (PROMEGA or QIAcube or QIAquick kit)
Gel Slice and PCR Product Preparation
Dissolving the Gel Slice
- Following electrophoresis, excise DNA band from gel slice in a pre-weighed 1.5 mL microcentrifuge tube.
- Add 10µL membrane Binding Solution per 10 mg of gel slice. We prefer not to vortex and we incubate at 50-65°C until gel slice is completely dissolved (∼10 min). Quick centrifuge.
Processing PCR reactions
For products above 40 pb
- Add an equal volume of Membrane Binding Solution to the PCR reaction.
Binding of DNA
- Insert the SV Minicolumn into Collection Tube.
- Transfer dissolved gel mixture or prepared PCR product to the Minicolumn assembly. Incubate at room temperature for 1 minute.
- Centrifuge at 16,000 x g for 1 minute. Discard the flowthrough and reinsert Minicolumn into Collection Tube.
Washing
- Add 700 µL Membrane Wash Solution (ethanol added). Centrifuge at 16,000 x g for 1 minute. Discard flowthrough and reinsert the Minicolumn into Collection Tube.
- Repeat Step 4 with 500 µL Membrane Wash Solution. Centrifuge at 16,000 x g for 5 minutes.
- Empty the Collection Tube and recentrifuge the column assembly for 1 min with the microcentrifuge lid open (or off) to allow evaporation of any residual ethanol.
Elution
- Carefully transfer Minicolumn to a clean 1.5 mL microcentrifuge tube.
- Add 30 µL Buffer EB (Qiagen). Incubate at room temparature for 1 minute. Centifuge at 16,000 x g for 1 minute.
- Discard Minicolumn and store at 4 or -20°C.
Quantification by Electrophoresis
- Gel : 1.5-2% agarose with BET added (5 µL BET for 100 mL TBE)
- 10 µL Quick-Load 1 kb DNA Ladder
- 2 µL LB + 3 µL DNA
Ligation
- 2 µL Ligase Buffer 10X
- Vv µL of measured [DNA]Vector in ng/µL (30 to 50 ng)
- Vi µL of measured [DNA]Insert in ng/µL
- Pure water QSP 20 µL
- 1 µL T4 DNA ligase at 400 000 U/mL concentration
- O/N at 16°C
Vi = (3 x [DNA]Vector x Vv x LenghtInsert) / ([DNA]Insert x LenghtVector)
Transformation
Use chemically lab-made competent cells or from a kit (DH5α, TOP10, Mach1)
- Defroze competent cells on ice during 5'
- Add 5 µl of DNA Ligation in 50 µL of competent bacterias (or 1 µL for the positive control puc19)
- Incubate 30' on ice
- Heat-shock the cells during 30" at 42°C without shaking
- Put 2' on ice
- Add 250 µL of pre-warmed SOC medium (42°C)
- Incubate 1h at 37°C under shaking (225rpm)
- Spin at 5.000rpm during 30"
- Remove 150 µL of supernatant
- Resuspend the pellet in the 150 µL left
- Spread on appropriate plates
- Incubate O/N at 37°C
LB and LBA
- LB medium: Dissolve 25mg/L of Luria Broth in purified water. Autoclave the solution. Store at room temperature and take care of any contamination
- LBA medium: Dissolve and gently mix with a magnetic barrel 40mg/L of Luria Broth Agar in purified water. Store at room temperature and take care of any contamination. To make some plates with this medium heat the solidified medium with microwave. Wait untill you can hold the bottle in your hand and then add the appropriate antibiotic to a final concentration of 1X. Dispend approximatively 20mL per plate in a sterile hood. Returned the plates and store them at +4°C.
1000X stock antibiotic
- Chloramphenicol: dissolve 30mg/mL of chloramphenicol in Ethanol 100%. Store at room temperature
- Tetracyclin: dissolve 12,5mg/mL of tetracyclin in a 50/50 Ethanol/water mix. store at -20°C
- Ampicillin: dissolve 100mg/mL of ampicillin in water. Filter the solution. Store at +4°C
- Kanamycin: dissolve 100mg/mL of kanamycin in water. Filter the solution. Store at room temperature
PCR Screening
Use of 8 clones of Ligation transformants for screening PCR
- One toothpick of each clone's colony per PCR tube
- Use toothpick to start 7.5mL O/N culture
After, add
- 12.5 µL Mix
- 0.5 µL Oligo F (10µM)
- 0.5 µL Oligo R (10µM)
- 11,5 µL pure water
- negative control : without clone's colony
- positive control
Store the tubes on ice waiting for PCR attains 95°C then put the tubes in the machine
LID 105°C
1. 95°C 5min
2. 95°C 30 sec
3. 55°C 30 sec
4. 72°C (1 min for 1kb)
5. go to : 2 rep : 29
6. sound : 1
7. hold : 4°C
Electrophoresis Purification of PCR
- 10µl of ladder 100 pb or 1 kb
- 4 µl of screening PCR
- Migration at 100V on 1,5% gel until 3/4 way
Sequencing
[http://institut.cochin.inserm.fr/rubric_recherche/Plates-Formes/sequencage_genomique/I18NFolder.2005-02-10.4781618697/page2/fr Sequencing COCHIN]
Promoter Characterization Plan
For theoretical consideration, see estimation of parameters
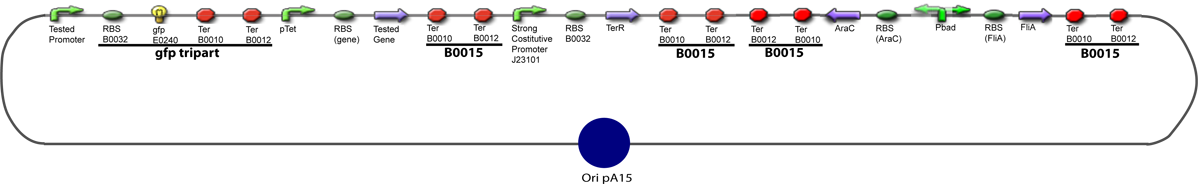
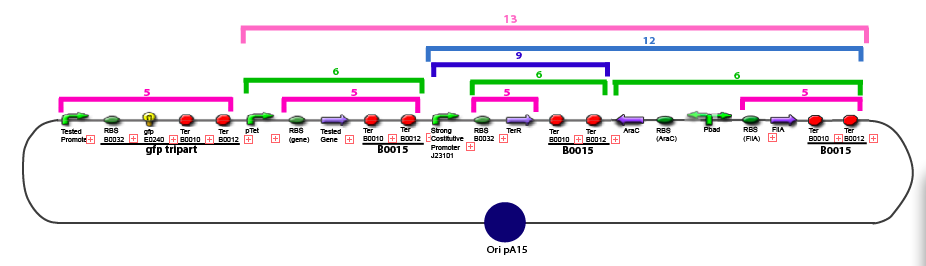
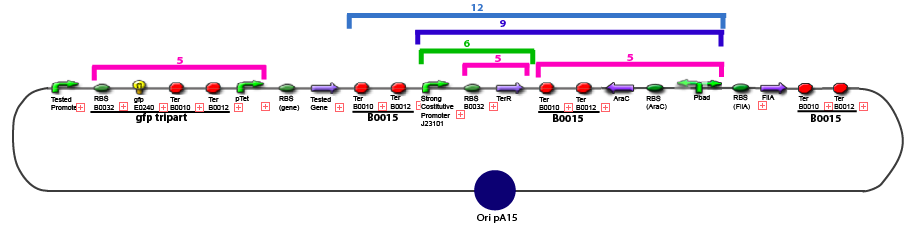
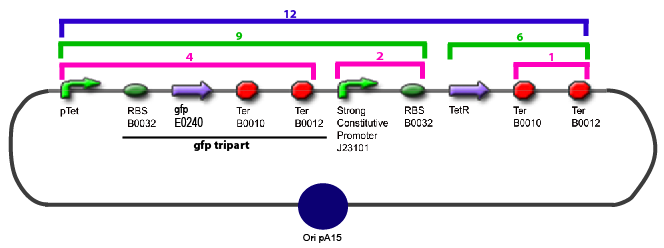
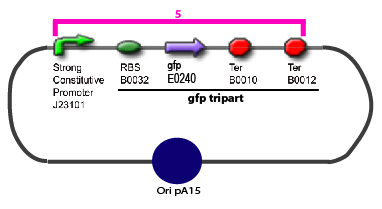
The same colour coded steps can be perfomed at the same time if elements needed are available
The order for treating the colours should of course be:
|
| Availability
|
| Promoters
|
| J23101
| yes
|
| pTet
| yes
|
| PflhDC
| ?
|
| PfliA
| ?
|
| PfliL
| ?
|
| PflgA
| yes
|
| PflgB
| yes
|
| PflhB
| yes
|
| Genes
|
| tetR
| yes
|
| gfp E0240
| yes
|
| ompR*
| ?
|
| envZ*
| ?
|
| fliA
| no
|
| flhDC
| yes
|
| Plasmids
|
| PSB3K3
| yes
|
| RBS
|
| B0032
| ?
|
| Terminators
|
| B0010
| ?
|
| B0012
| ?
|
| Bacterial Strains
|
| Top10
| yes
|
| FliA -/ FlhDC -
| ask Ariel
|
| FlgM -
| ask Ariel
|
This table contains the promoters we need to characterize, the transcription factors whose effect on the promoter we want to test, and the plasmid we want to obtain in order to carry out each characterization
| Promoter
| Promoter
Availability
| TF gene
| TF gene
Availability
| Done/Not done
|
| J23101
| yes
|
|
|
|
| pTet
| yes
|
| ?
|
|
|
|
| tetR
| yes
|
|
| None
| ?
| gfp E0240
| yes
|
|
| PflhDC
| ?
| ompR*
| ?
|
|
| PflhDC
| ?
| envZ*
| ?
|
|
| PflhDC
| ?
| fliA
| no
|
|
| PfliA
| ?
| flhDC
| yes
|
|
| PfliA
| ?
| fliA
| no
|
|
| PfliL
| ?
| flhDC
| yes
|
|
| PfliL
| ?
| fliA
| no
|
|
| PflgA
| yes
| flhDC
| yes
|
|
| PflgA
| yes
| fliA
| no
|
|
| PflgB
| yes
| flhDC
| yes
|
|
| PflgB
| yes
| fliA
| no
|
|
| PflhB
| yes
| flhDC
| yes
|
|
| PflhB
| yes
| fliA
| no
|
|
Protocol to make competent bacteria
1. Use non competent bacteria (ex: MG1655) stocked in 1.5 mL LB (20% Glycerol): put a toothpick in the 1.5 mL stock tube and then place it in a 50 mL Falcon with 5 ml LB medium. Over Night culture at 37°C / 300 rpm
2. 1/100 dilution in LB medium QSP 50 mL in an erlenmeyer of 250 mL
3. Culture at 37°C / 300 rpm untill OD600 reach 0.6
4. Fast cooling at +4°C by gently shaking the erlen in ice
Before: prepare CaCl2 0.1M.
- Add 5,56 g in 500 mL H2O (Gibco)
- dissolve the CaCl2 by mixing the suspension with the help of a magnetic stirrer
- Filter the solution with a cell-culture unit of filtration
- Aliquotes the filtered solution: 25mL in a 50 mL Falcon. Storage at +4°C
5. Use pre-cooled centrifuge at +4°C. Centrifuge 50 mL of the culture in 50 mL falcon: +4°C / 5 min / 5000 rpm
6. Discard supernatant by inverting the tube, and resuspend the pellet with 1 mL of cold CaCl2 and mix gently the suspension by up and down
7. Add cold CaCl2 QSP 20 mL and incubate 30 min / +4°C
8. Centrifuge the suspension : +4°C / 5 min / 5000 rpm
9. Discard supernatant by inverting the tube, and resuspend the pellet with 1 mL of cold CaCl2 and mix gently the suspension by up and down
10. Transform or freeze the competent cells. Freeze the competent cells in 50 µL aliquots in the 0.1M CaCl2 medium with 15% glycerol.
11. After transformation, prepare a Glycerol Stock or/and use the transformed bacteria to study the doubling time of the bacteria population
Study of the doubling time of the bacteria population
- B1 Vitamin 0.1%
- Uracile 0.2%
- MgSO4 2 mM
- CaCl2 0.1 mM
- Glycerol 0.4%
- Casamino acids 0.1%
- M9 minimal salts medium 1x
- H2O QSP 200 mL
- Filter the medium with a cell-culture unit of filtration
- Unfreeze strains from the glycerol stock containing or not the plasmid with a contitutive promoter and a fluorescent reporter gene
- Put a sterile tip in the 1.5 mL stock tube and then place it in a 50 mL Falcon with 5 ml of the medium above. Over Night (16h) culture at 37°C / 300 rpm
- Measure the OD600 (Linear Zone Measurement <1.5)
- Dilute the culture medium in the same new medium to obtain an approximative OD600 of 0.01 (Vfinal= 50 mL in a 250 ml erlenmeyer)
- Incubate à 37°C / 300 rpm and measure the OD600 every 20 min
- Determine the doubling time population and compare the strains containing or not the plasmid with a contitutive promoter and a fluorescent reporter gene
|
 "
"






