 "
"
Team:Illinois/Antibody GPCR Fusion
From 2008.igem.org

| Home | Team | Project | Notebook | Research Articles | Parts | Protocols | Pictures |
Contents |
Core Team Members
Dave Luedtke, Bobak Hadidi, Kiruthika Selvadurai, Namita Bakshi
Project Abstract
G protein-coupled receptors, or GPCRs, are transmembrane receptors that sense extracellular objects on the scale of small molecules to large proteins. "The importance of GPCR systems is illustrated by the fact that 30% of all clinically prescribed drugs function as GPCR agonists or antagonists" [2]. Activation of the GPCR by ligand binding begins a signal transduction pathway that ultimately results in the transcriptional activation or repression of one or more genes. The signal is transduced with G-proteins, which are signalling proteins that associate with GTP and GDP, as well as kinase cascades. Yeast cells are known to utilize two GPCRs signal transduction pathways, one to detect the presence of glucose and the other to initiate mating. We hope to engineer the well-characterized mating pathway to produce a colorimetric change in the cell upon detecting a novel molecule-- a surface protien of some water-borne pathogen, or possibly a toxin secreted by such a water-borne pathogen.
Specific Plans, Supplies, and Protocols
Current Plan
We plan to use a steric hindrance mechanism for indirect detection of the presence of the cholera toxin. Rather than modifying the GPCR's target ligand, the addition of the Antibody on the N-terminus of the protein would act as a gate to allow or block binding in the original receptor domain. In this method, the addition of the AB on the end of the GPCR could conceivably:
- Block the normal binding site for the alpha factor, unless the AB was bound. After adding a LacZ or GFP gene under the control of the FUS1 promoter, if the yeast was not fluorescing, then we would know toxin was present.
- Vice versa; that is, if the AB was bound by the toxin it would block the binding of the natural ligand to the GPCR so the signal would not be induced. Thus, once production of LacZ or HIS or GFP halts we could know toxin was present.
In either case we would modify the original GPCR gene with spacer dna attached to the antibody gene. After transforming the cells with that plasmid as well as the Fus1-GFP plasmid, we would test the mechanism by adding purified alpha factor to the solution.
Alpha factor has the effect of causing cell cycle arrest on MATa yeast, through the very same pathway. This would obviously have adverse affects on our cell cultures; in order to prevent this, we will have to knockout the Far1 gene.
Once we know the original working parts are in order, rounds of directed evolution, selecting for candidates showing the steric mechanism detailed, would be conducted. DE should be more focused on the AB chain, leaving the receptor mostly untouched.
It seems the most important variable, outside of the directed evolution, is the length of the spacer dna. It must be long enough so that the two fused proteins can fold without interfering with each other, as well as the correct length so that the AB may cover the ligand binding domain. I think alpha factor typically binds to the 3rd extracellular loop of GPCR.
Steps:
- Obtain Yeast lacking Ste2, alpha factor, plasmids, and cholera toxin
- Knockout FAR1 gene
- Add GPCR-Antibody fusion protein plasmid
- Add FUS1-Reporter (GFP or LacZ) plasmid
- Use directed evolution iterations on ABSte2 plasmid
- New yeast strain that detects for cholera toxin through the pheromone pathway
Preventing Cell Cycle arrest by mutating the FAR1 gene
- This gene is situated on chromosome X; it is a "Factor ARrest" gene
- FAR1 gene is induced and cell-cycle arrest begins with the secretion of peptide pheromones a and alpha by haploid yeast cells; arrest occurs in G1 phase
- Creating null mutants:
- Two substitutions in FAR1 lead to the far1-D1 mutation--glycine 646 and proline 671 are replaced by aspartic acid and leucine, respectively (Figure 3). This null mutant can still induce the signal transduction pathway as in the wild-type, but is unable to halt the cell-cycle at the G1 phase when exposed to mating pheromone.
- [1]<--- Scroll down to figure 3. The Glycine 646 and proline 671 that have to be mutated are labeled in blue.
Homologous Recombination Method [2]:
- Know the DNA sequence of the gene we want to replace
- Construct a DNA sequence with gene of interest (GOI) which has extra DNA on either side of GOI which is identical to the DNA on the genome (The picture on the website makes it clearer) + attach antibiotic resistance gene
Problem: The mutated FAR1 gene is different from the original FAR1 gene by 2 nucleotides, which could mean that recombination could occur anywhere. We can also replace it with the URA3 gene instead.
Protocol for Homologous Recombination [3] - We can use Basic Protocol 2 or 3
Sequence of FAR1 [4]
Sequence for URA3 gene [5]
Fusion Protein Plasmid
- Linker DNA must be between AB and Ste2 proteins so they can fold properly
- Ensure fusion protein sent to the plasma membrane
- "Prenylation" adds hydrophobic molecules to proteins, usually with the goal of localizing them to the cell membrane. In Yeast this can be accomplished by addition of the CaaX amino acid sequence 4 aa's from the C terminus of the protein; where C stands for cysteine, a is any aliphatic amino acid, and the identity of X determines which enzyme acts on the protein; List of Aliphatic AA's.
- The yeast a factor is shown to contain the CaaX motif. Amino Acid selection for Caax in yeast LINK
- We plan to extract Ste2 gene (~1200 base pairs) from the yeast genome and put it on a plasmid Coding Sequence
Outline:
RE1: Eco RI 5' G^AATTC
RE2: Sac I 5' G^AGCTC
RE3: Xba I 5' T^CTAGA
RE5: Hind III 5' A^AGCTT
== plasmid ==== RE1 == PGK promoter == RE2 == == more restriction sites which include REX/Y (may be RE2/3) for antibody gene == == RE3 == Ste2 == RE4 == PGK terminator == RE5 ==== plasmid ==
Integration of Fluorescence Reporter Controlled by FUS1 Promoter
Scroll down to the 'Materials and Methods' [6]
Sequence of FUS1 [7]
Integrate or Plasmid
We are currently considering direct integration of the FUS1-Fluo.Prot. fusion into the yeast genome, which seems to be the standard for this type of thing.
Basically the target gene would be integrated by flanking it with complementary bases so the entire DNA fragment would undergo homologous recombination with the appropriate section of the yeast chromosome, replacing the original DNA.
Our required materials include 4 Biobrick parts and 5 regions of DNA cloned out of the yeast genome (flanking where we would integrate).
Biobricks:
- EYFP .................... BBa_J63001 ........ 744 bp
- ADH1 Terminator ......... BBa_J63002 ........ 225 bp
- HIS3 Marker ............. BBa_Y1006 ......... 660 bp
- includes its own promoter and terminator
- Biofusion Plasmid ....... BBa_J63010 ........ >4k bp
- includes ampicillin resistance gene
- EcoRI site found in the sequence at base pair 1991.
- XbaI site found in the sequence at base pair 2006.
- SpeI site found in the sequence at base pair 2012.
- PstI site found in the sequence at base pair 2026.
- NotI site found in the sequence at base pair 1997.
- NotI site found in the sequence at base pair 2019.
Yeast DNA:
- FUS1 promoter with ~250 amino acids of FUS1 gene
- 1kb region downstream of FUS1 (~1000 bps)
- Ste2 gene (1296 bps)
- PGK Promoter (657 bps)
- PGK Terminator (288 bps)
Directed Evolution/Mutagenesis
- Error Prone PCR in vivo
- Site Directed Mutagenesis
Protocols
- Preparation of Yeast Media [8]
- Basic Protocol 1: Growth in Liquid Media[9]
- Rapid Isolation of Yeast Chromosomal DNA [10]
Possible Experiments
- Test the effect of alpha factor on GPCR pheromone-deficient (Ste2) yeast strain
- Compare growth of control plate versus growth on plate+alpha factor
- Create Ste2-plasmid using restriction enzymes
- Add plasma membrane shipping tag sequence (we need to find it for yeast)
- Create plasmid with GFP or LacZ under control of Fus1
- Add to regular yeast strain with MATa and MATalpha type, should see regular fluorescence.
- Add to Ste2 deficient strain, shouldn't see any fluorescence.
- Add to chromosomal Ste2 deficient strain with unmodified Ste2 plasmid, should see regular fluorescence.
- Create Toxin Antibody Plasmid by combining heavy and light chain sequences
- Need some way to test if it folds properly/works, maybe use protein scaffold idea.
- Test transformation of Ste2 deficient strain with Ste2-plasmid
- Growth should be comparable to strains with Ste2 in genome
- Modify Ste2-plasmid by adding linker dna and complete antibody dna (ABSte2)
- Have to find appropriate length/sequence for linker dna so that receptor folding and antibody folding once translated don't interfere with each other.
- Transform deficient and wild type strains with ABSte2
- Test effects of alpha factor on strains. See if AB interferes with signal transduction.
Previous Plan
In theory, this is how we will progress:
- Create a fusion protein that links an antibody against cholera toxin to the Ste2 GPCR of S. cereviviae, the pheremone response GPCR.
- 1)Find the sequences of the GPCR [11] and the antibody. Did someone already find the sequence of the antibody? [12] <--- We can buy the antibody to the beta subunit here.
- 2)Select site of fusion
- Cys187 in EL2 and Cys110 in TM3: highly conserved cysteines found in most GPCRs
- "extracellular loop 2 has extensive contacts with the other extracellular domains and also plunges down into the transmembrane bundle, contacting the retinal ligand." If we add the antibody here, will it interfere with the proper folding of the surface protein? [13]
- "the first extracellular loop is important in ligand-mediated activation of the receptor, whereas the second loop functions as a negative regulator of receptor activation and serves to stabilize the inactive state of the receptor" [14]
- Possible sites for fusion of antibody: Tyr101 through Gln135 in EL1 domain (because they did not have major effects on binding and signal transduction when they were mutated to Cysteine)
- Also, we want to add the antibody at a point where the unbound state (of the GPCR) is in contact with the solvent: residues 101 and 106
- "mutation of residues Leu102, Asn105, Ser108, Tyr111, and Thr114 to cysteine resulted in receptors that could effectively bind pheromone, but they were partially (Leu102 and Gln135) or severely (Asn105, Ser108, and Tyr111) compromised with respect to signal transduction." [15]
- 3)Have the gene sequenced--This will be a lengthy gene. We might be better off somehow obtaining a gene containing the natural GPCR (perhaps from a research group) and inserting the gene of the antibody.
- Professor Chris Rao explained to us an online tool (it was some company) for determining what restriction enzymes and where in the gene they could act to aid us in this.
- One yeast Ste2 gene was sequenced to be ~1.5kb, including restriction sites for EcoRI and BamHI [9].
- Express the antibody/GPCR fusion protein in yeast that lack the wild type receptor.
- Found 6 strains (type "ygk ste3|ste2" without quotes into the text search) of yeast of interest from the Yeast Genetic Stock Center, 2 of mating type a, 2 of mating type alpha, and 2 that are diploid (both). Three of each type have their Ste2 gene knocked out; the other 3 likewise have their Ste3 gene knocked out. Each costs about $150, in addition the media for their growth conditions is listed, WOW!
- MATa expresses Ste2 (the alpha-factor receptor) while MATalpha expresses Ste3 (the a-factor receptor) [1]. We should thus either order MATa without Ste2 or MATalpha without Ste3.
- Which mating type would better suite our purposes?
- The gene will most likely be on a plasmid. We may have to integrate the gene into the yeast's chromosome, however.
- The size of the gene may be substantial, if it turns out to be too large for a plasmid, integration into the yeast chromosome may be the way to go (hopefully not).
- [9] cites that a form of the Ste2 gene has been carried by a vector into yeast, size should not be a problem unless the antibody module is large.
- The size of the gene may be substantial, if it turns out to be too large for a plasmid, integration into the yeast chromosome may be the way to go (hopefully not).
- Found 6 strains (type "ygk ste3|ste2" without quotes into the text search) of yeast of interest from the Yeast Genetic Stock Center, 2 of mating type a, 2 of mating type alpha, and 2 that are diploid (both). Three of each type have their Ste2 gene knocked out; the other 3 likewise have their Ste3 gene knocked out. Each costs about $150, in addition the media for their growth conditions is listed, WOW!
- Measure the activation of the GPCR by the toxin (and by the natural pheremone) using a reporter gene.
- Several studies have used the FUS1 promoter in conjunction with HIS3 selection. The FUS1 promoter seems to be a good choice for our purposes.
- Here's a recombinant plasmid we will need to contruct:
[16] a. the FUSI promoter isolated from Saccharomyces cerevisiae;
b. DNA encoding approximately the first 254 amino acids of said FUSI gene; FUS1 gene in Yeast (FASTA format): >FUS1_YEAST MVATIMQTTTTVLTTVAAMSTTLASNYISSQASSSTSVTTVTTIATSIRSTPSNLLFSNVAAQPKSSSAS TIGLSIGLPIGIFCFGLLILLCYFYLKRNSVSISNPPMSATIPREEEYCRRTNWFSRLFWQSKCEDQNSY SNRDIEKYNDTQWTSGDNMSSKIQYKISKPIIPQHILTPKKTVKNPYAWSGKNISLDPKVNEMEEEKVVD AFLYTKPPNIVHIESSMPSYNDLPSQKTVSSKKTALKTSEKWSYESPLSRWFLRGSTYFKDYGLSKTSLK TPTGAPQLKQMKMLSRISKGYFNESDIMPDERSPILEYNNTPLDANDSVNNLGNTTPDSQITSYRNNNID LITARPHSVIYGTTAQQTLETNFNDHHDCNKSTEKHELIIPTPSKPLKKRKKRRQSKMYQHLQHLSRSKP LPLTPNSKYNGEASVQLGKTYTVIQDYEPRLTDEIRISLGEKVKILATHTDGWCLVEKCNTQKGSIHVSV DDKRYLNEDRGIVPGDCLQEYD c. DNA encoding a protein or a polypeptide of interest (Could be His3), the DNA positioned so as to be under the control of said FUSI promoter;
d. a two micron autonomously replicating sequence;
- The usual response of haploid yeast cells to the mating pheromone leads to the activation of Pheromone Responsive genes and also cell cycle arrest. For the phenotype to be removed and the cells to continue growing when the pheromone response pathway is activated, the FAR1 gene should be deleted.
- Use site specific directed evolution to increase the effectiveness of the new GPCR.
- [17]Methods of site-directed mutagenesis, including non-PCR techniques
- [18]Protocol for site-directed mutagenesis using PCR
- Altered-States Method[19]:
- 1. Start with a plasmid carrying a defective selectable marker (e.g. Amp)
- 2. Link the mutation you are making elsewhere in the plasmid to a correction of the defective selectable marker.
- 3. Select for correction of the selectable marker, and you are likely to also find plasmids with your specific mutation introduced as well
- QuikChange Method[20]
- Gene of interest in plasmid, but it is modified (bp's added, removed, substituted), DNA is denatures and primers anneal to both strands, there are "staggered nicks" where mutation is, Dpn I added (digests methylated DNA), parent DNA is methylated but new mutated DNA is not, result is that only new mutant DNA (see below, brown region w/ green x's) is transformed in bacteria cell
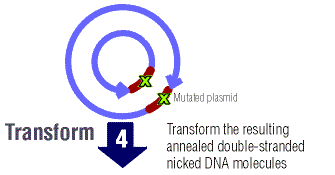
Alternatively, we might begin looking into completely replacing the yeast receptor with a mammalian or novel receptor with an affinity for our target protein. If we go down this route, we will also have to replace the G-alpha subunit with a chimeric one that has affinity for both the new receptor and the yeast G-betagamma complex. These have already been synthesized/cloned/will be fairly easy to procure, and may or may nor be receptor specific. This method is the major procedure for researching GPCRs today, typically used in pharmaceutical research -- knock out the pheromone GPCR while leaving the endogenous signal pathway/reporter intact, then introduce the uncharacterized GPCR of interest (with its G-aplha). In our case, we would not be studying an unknown GPCR to find what its target ligand or purpose is; instead we would simply already have one that we had/would evolve using mutagenesis to have an affinity for the toxin.
To Research
- Effect of constitutive expression of alpha factor on MATa yeast cells. While they could still divide by budding, the signal pathway causes cell growth arrest in G1; is this a problem??
- How to knockout the gene responsible for growth arrest when alpha factor is present.
- Cell Wall Issue
- "The potency of some ligands might be reduced in yeast compared with mammals [for receptors taken from mammalian cells]. Using Gpa1–Ga chimeras probably explains part of this discrepancy [original yeast G-alpha subunits have low affinity for the new receptors and the mammalian G-alpha has low affinity for yeast G-betagamma, we will bypass this problem altogether by fusing the antibody to the original yeast receptor], but another factor might be the ability of ligands to penetrate the yeast cell wall. Potencies for small ligands are similar in yeast and mammals, those for intermediate ligands are about two orders of magnitude less in yeast, and large ligands such as chemokines (polypeptides with 70–80 residues) are often unable to activate receptors when applied exogenously to yeast. The fact that smaller versions of these ligands can be more efficient agonists suggests that accessibility is an issue."[1]
- Antibody Sequence (we have)
- Receptor Sequence (we have)
- Alpha Factor Sequence - 13 AAs (WHWLQLKPGQPMY)
- Sources of yeast strains deficient in specific genes (we have)
- Cholera Toxin or possible target protein
G Proteins and their Receptors

G proteins are comprised of 3 subunits, collectively known as a heterotrimeric or large G protein. The individual pieces are Gα, Gβ and Gγ. When the appropriate molecule is bound to the receptor, the GDP bound to the Gα unit becomes GTP. More conformational changes are induced causing Gβγ and Gα to dissociate. Depending on the signal cascade, either complex, Gα-GTP or Gβγ may now induce the appropriate linked signal pathway.
As opposed to other signal receptors, the ligands that bind GPCRs typically bind within the transmembrane domain. When the receptor is bound by its ligand, it shifts shape, activating the G protein (by allowing the exchang of GDP for GTP). Also, G protein complexes may or may not be coupled with the receptors, instead, if they happen across a receptor with its activating ligand bound, the G protein will be activated (GDP -> GTP) and induce its signal cascade. (wikipedia).
Literature Research and References
[1]Functional analysis of heterologous GPCR signaling pathways in yeast
[2]http://www.nature.com/embor/journal/v2/n7/full/embor385.html
Ste2 information, includes sequence:http://www.bio.davidson.edu/Courses/genomics/2002/Statler/STE2.htm
[9] Ste2 Protein
2 GPCR pathways in yeast: 
| Home | Team | Project | Notebook | Research Articles | Parts | Protocols | Pictures |