|
Home
The Team
Project Report
Parts
Modeling
Notebook
Safety
CoLABoration
|
_meetings
- Apr. 24th 2008
- Apr. 30th 2008
- May 8th 2008
attendees
Philipp Mappes, Wolfgang Schamel, Kilian Bartholomé, Katja Arndt, Kristian Müller, Dario Hermida Aponte, Normann Kilb, Robert Gawlik, Simone Weber, Veronika Götz, Kristina Brückner, Kathrin Pieper, Michael Kneib, Moritz Busacker, Sabine Jägle
How to organize the project?
The DNA origamis shall be tested on the established T-cell system. Then we will try to establish an artificial system. Extracellular fragments could be the EGFR or Fas for example. Since the Fas receptor is involved in apoptosis, signaltransduction could be detectable by cell death. Further systems for detecting signaltransduction could be NF-kappaB, the MAP-Kinase-pathway, the Jun-Fos-system or splitenzymes.
DNA-origami structure
How expensive will the synthesis of the NIP coupled Oligos be? Phillip and Michael will contact the company and ask for an calculation of costs. NIP can be ordered by the company Biosearch technology.
Controls for the DNA origamis
- We should produce a DNA origami without NIP-coupled Oligos to check if there also a cell binding occurs (negative control)
- To check the calculated distances between the NIP's on the origami a origami with fluorophores could be made. By using FRET the distance between the oligo's can be estimated.
- May 23th 2008
- Jun 2th 2008
attendees
Sabine Jägle, Normann Kilb, Robert Gawlik, Kristian Müller, Philipp Mappes
We have talked about...
- To ensure an optimal NIP-scFV-Anti-NIP binding a longer NIP-Linker should be used
- Calculated distances between the NIP's are 6-7 nm.
- Integration of the TCR in the model to achieve a comparison of the sizes
- Searching a suitable transmembraneregion for the artificial receptor
- Calculation of costs for 10 Oligos wirh NIP coupled to the 5' or 3'-end: 3000,-US$ by unknown coupling efficiency
- => Ordering presumably by PURIMEX
- Jun 13th 2008
attendees
Michael Kneib, Robert Gawlik, Philipp Mappes, Simone Weber, Kristina Brückner, Sabine Jägle, Kathrin Pieper
Ordering of NIPs
Assembly of the DNA origami wiht the coupled NIPs was discussed and decided:
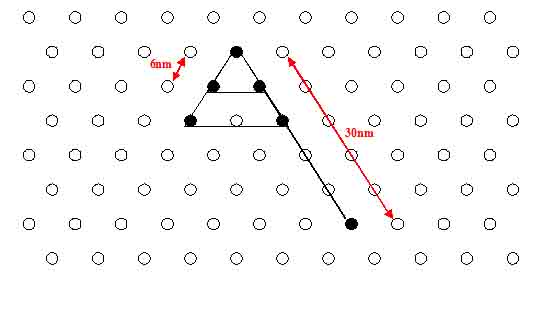
Question: What are the exact distances between the DNA-helices? --> Rothemund
The next steps
- searching for literature of TCR-Clustering
- searching for literature transmembranhelices, signalesequence
- planning the artificial system with antibodyfragment, transmembraneregion, splitenzymes, enzyme assay
- EGFR as further testsystem
- 16. Juni 2008 10.00h
Vorbeireitung der Präsentation für Dienstag 12.00h
- 17. Juni 2008 12.00Uhr
Besprochenes
- Bis Freitag werden nach Rücksprache mit Schamels Doktorandin die NIP-gekoppelten-Oligos bestellt --> Phillip
- Die Vektoren für die Klonierungen zusammentragen (Dina/Christina: Vektor mit CMV-Promotor) --> Kathrin
- Schamels Versuche zum TCR "nachkochen" --> Terminabsprache mit der Doktorandin
- Klonierungsstrategien entwerfen (dabei IGEM-Schnittstellen-Vorgaben beachten)
- Literatur-Recherche Transmembranhelices WF DeGrado
- Programm für Klonierungen: Emboss (emboss.org)
- 23. Juni 2008 12.00Uhr
Anwesend
Robert Gawlik, Philipp Mappes,
Michael Kneib, Kathrin Pieper, Sabine Jägle, Kristina Brückner, Simone Weber
Besprochenes
·
Oligos sind bestellt und sollten in der Woche vom 10.Juli
geschickt werden.
·
Transmembran-Region:
o
Literaturrecherche zur Transmembranregion
o
Welche Transmembranregionen können benutzt werden z.B. EGF-R
o
Signalpeptid?!
o
Paper: Erythropoitetin Receptor Activation by a Ligand-Induced
Conformation Change
·
synthetische Transmembranregion:
o
Aus welchen Teilen soll der synthetische Rezeptor zusammen gebaut
sein?
·
Vektoren:
o
Genkarten für Vektoren müssen gesammelt werden
o
Vorhandene Vektoren müssen mit den IGEM-System(Freiburg2007-WIKI
Seite) verglichen werden
o
evtl. suche nach geeigneten Vektoren
Aufgaben
·
Literaturrecherche Transmembranregion/Signalpeptid : Simone, Kristina,
Sabine
·
Vektorsequenzen : Philipp, Kathrin,
Michael
·
Einrichten unserer Gruppe auf offizielle Wikiseite :Robert
- 3. Juli 12.00 Uhr
Teilnehmer
Robert Gawlik, Michael Kneib, Kathrin Pieper, Simone Weber, Daniel Hautzinger, Sabine Jägle, Philipp Mappes, Kristian Müller
1. Vektoren
Philipp und Kathrin haben sich in GCG eingearbeitet. Dabei hat sich gezeigt, dass in der Fab Fragment Sequenz eine IGEM Standard Schnittstelle befindet. In dem Vektor von Christina befinden sich 7 Schnittstellen.
Es bieten sich die Möglichkeiten der klassischen Mutagenese oder der Gensynthese. Von Geneart gibt es das Gensyntheseangebot 20 Cent pro Base. Dies gilt allerdings nur für 2500 Euro, anschließend gilt der Preis von 50 Cent pro Base. Falls wir die Gensynthese machen wollen, sollte man bei Geneart anfragen, ob die Vektoren, die sie verwenden auch keine IGEM (EcoRI, XbaI, SpeI, PstI) und keine von unseren Schnittstellen (NgoMIV, AgeI) haben.
Die restlichen Sequenzen, die noch gefehlt haben, hat Mahima inzwischen im richtigen Format geschickt, sodass diese Sequenzen nun auch noch auf die Schnittstellen untersucht werden können (Kathrin und Philipp?).
Man könnte auch noch mal auf der bei den IGEM parts nach Vektoren mit CMV Promotor schauen.
2. Konzentration der DNA Struktur
Daniel hat eine Konzentration von 20 nM berechnet, die eventuell auch auf 200 nM erhöht werden könnte . Laut Mahima ist aber mindestens eine Konzentration von 2 µM nötig. Eventuell können wir eine geringere Konzentration einsetzen, allerdings können wir dann wahrscheinlich keine Unterschiede mehr detektieren, ob 2, 3 oder 4 NIP's gebunden haben.
3. DNA Strukturen
- Kontrolle ob DNA mit NIP's an die Zellen bindet. Dafür DNA Struktur mit 4 NIP's und 2 Fluorophoren. Den Nachweis der Fluoreszenz als Beweis für die Bindung der DNA Struktur an die Zellen. Siehe auch Konfokale Mikroskopie.
- Negativkontrolle. DNA Struktur ohne NIP's, mit 2 Fluorophoren. Nachweis dass diese Struktur alleine nicht zu einer Stimulation des T-Zellrezeptors führt.
- Michael, Simone und Daniel machen M13 Phagen Prep und den Oligopool.
4. Nachweis der T-Zellrezeptor-Stimulation
- Ca-Messung: Bei 530 nm. Gerät im MPI ist allerdings kaputt?
- GFP Marker: Genexpression ist nach 12-18 h nachweisbar. GFP positive Zellen werden mit dem FACS bestimmt. Eventuell wäre es möglich ein FACS in der Uniklinik zu verwenden. Kathrin erkundigt sich bei Dr. Eibel.
5. Konfokale Mikroskopie
- Im MPI muss man zuerst ein Trainingsprogramm absolvieren, worauf man allerdings auch 3 Wochen warten muss
- Wir könnten das konfokale Mikroskop im Fischbach Labor nutzen. Da stehen die Wellenlängen 488nm und 568nm zur Verfügung.Ansprechpartner ist Gerit.
6. Allgemeines
- Rezepte von Puffern usw auf's Wiki schreiben
- Wenn man etwas Neues in Erfahrung gebracht hat Rundmail schreiben, später dann aber auch ins Wiki
- Laborjournal wird chronologisch geführt
- Anlegen eines Registers z.B. auf der ersten Seite des Laborbuchs. Hier kann man dann noch mal ausführlicher beschreiben was genau in den Eppi's drin ist
10. Juli 2008 12:00 Uhr
Teilnehmer:
Simone Weber, Sabine Jägle, Kathrin Pieper, Phillip Mappes,
Norman Kilb, Robert Gawlik, Daniel Hautzinger, Michael Kneib, Kristian Müller,
Kristina Brückner
1. Sicherheitseinführung
> interne Notrufnummer: 2000
2. Stand der Dinge
- M13-Phagen DNA wurde geprept
- inzwischen sind alle Oligos eingetroffen --> Pool
wurde hergestellt indem alle drin sind, außer denen, die Fluoreszenzmarkiert
werden oder NIPs tragen
- mit DNA-Origamie wurde begonnen --> AFM-Betrachtung
11.07.08
- ersten Oligos müssen nachbestellt werden (~ 50€)
3. Lösung für Konzentrationsproblem
- Ca2+ - Einstrom direkt unter dem
Fluoreszenzmikroskop anschauen
- welcher Farbstoff hier verwendet werden soll --> wird
am Freitag, 11.07. besprochen
4. Überlegung für einfaches synthetisches System
- könnten bereits vorhandenen Vektor nutzen, der
β-Kette des TCR + das Fab-Fragment enthält
- auf cytoplasmatische Seite einfach noch β-Lactamase
dran synthetisieren
- zeigt nur, das die NIPs die β-Ketten zusammen und
dadurch die die β-Lactamase aktiviert wird
5. Substrat für β-Lactamase
- muss demnächst bestellt werden
- welches --> Norman informiert sich
6. Protokolle für Transformationen
- in Kristians Labor vorhanden
7. Positiv Kontrolle für Transfektionen
- Norman kümmert sich darum
8. Suche nach weiteren Vektoren
- bei Toolbox Freiburg nachfragen
9. zweites System für Multimerisierung mit anderen
Antikörpern
- Antifluorescin-Antikalin (Arne Skerra) -->
Publikation in Literatur!
- Andreas Blukthuhn (?) --> Dapin (> Kristian kümmert
sich darum)
10. intrazellzläre Domäne des EGF-R
- <a href="http://www.ebi.ac.uk">www.ebi.ac.uk</a>
--> SRS --> Uniprot
- <a href="http://www.expasy.ch">www.expasy.ch</a>
--> swissprot
- Michael kümmert sich darum
16 Juli 2008 12.00h
Teilnehmer:
Robert Gawlik, Michael Kneib, Kathrin Pieper, Simone Weber, Moritz Busacker, Sabine Jägle, Philipp Mappes, Kristian Müller
1. DNA-Origami:
- nach geglücktem 1. Ansatz jetzt der Versuch mit DNA:Oligos = 1:5 (anstelle von 1:20 wie bisher)
- RPMI-Medium oder Ringer-Solution imaging-geeigneter?
- Herstellung von Fluorophor-Origami für erste Imaging-Versuche; Fluoreszenz-Mikroskopie im MPI oder Fischbach-Labor?
- Stabilität des Produktes (Transport etc.)?
Vektoren / Synthetisches Rezeptorsystem:
- nach wie vor noch kein uneingeschränkt nutzbarer Vektor verfügbar; evtl. vom Ljubljana-Projekt 2007 (CMV-Promoter als Modul)?
- deshalb evtl. Umweg über "Zwischenlagerungs-Vektor"
- Planung und Bestellung folgender Parts:
-Fab-Singlechain
-Signalsequenz für die Membranintegration?
-Transmembranregion?
-evtl. Split-Fluorophor?
- Integration von Kurzlinkern in die einzelnen Parts?
- für eventuelle EGFR-Parts Rücksprache mit Tillmann Brunner, ZBSA (downstream-signaling)
23 Juli 2008 12.00h
Teilnehmer:
Robert Gawlik, Philipp Mappes, Kathrin Pieper,Sabine Jägle,
Simone Weber.
EGF-Rezeptor (EGFR):
- EGFR als Transmembrandomäne sinnvoll da er sehr
groß ist.Auf der T-Zell-Oberfläche befinden sich
viele große Moleküle, diese sind viel
Größer als die Rezeptoren. Bei einem kleineren
Rezeptor wäre es möglich, dass die großen
Moleküle den Rezeptor überlagern und die Origamis
nicht binden können.
- Der EGFR könnte als ganzes System genutzt werden,
an dem die single chain über einen langen Linker gebunden
wird. So werden Moleküle weit genug heraus gebracht um
über andere Moleküle herauszuragen.
- Drei mögliche Klonierungsvorschläge:
1)EGFR + Single chain + 1.
Hälfte der beta-Lactamase
2)EGFR + Single chain + 2.
Hälfte der beta-Lactamase
3)vollständige EGFR (mit single chain): könnte man mit EGF die Aktivierungdes EGFR messen (welches read out?)
Fura 2/ Indo-1:
- können als Farbstoff für Ca2+ Farbstoffe
verwendet werden.
- wir benutzen Fura 2AM -> wird auf die Zelle gegeben,
aufgenommen, in den Zellen spalten Proteasen den AM- teil ab. Der
Farbstoff kann dann als gespaltenen Form die Zelle nicht mehr verlassen.
- gemessen werden Emissionswerte in einem so genannten
Ratiomeasurement, d.h. es wird bei 2 Wellenlängen gemessen und
das Verhältnis daraus berechnet.
Aug. 7th 2008
Teilnehmer:
Robert Gawlik, Philipp Mappes, Normann Kilb, Sabine Jägle,
Kristian Müller, Moritz Busaker.
- problem: Where is the next Ca2+ measurement?
- Take data of the Ca2+-measurement from Prof. Schamel. Titration to get the right concentration?
- pictures from measurement should be handled with "Zeiss Software" (Sabine)
- Carry over T-cells from Mahima (MPI) to kristians lab (Sabine)
- ATG-Biosynthetics synthesizes all parts in a vector without unintentional iGEM restrictionsites
- Invitrogen CCF4AM and CCF2AM for bla detection = LiveBLAzer™
- CCF2AM/CCF4AM 5mg 13.910.00 €
- CCF2AM Loading Kit 200 μg 815.00 €
- LiveBLAzer™-FRET B/G Loading Kit 200μg 690.80€
brochure for further information [http://tools.invitrogen.com/content/sfs/manuals/liveblazer_FRETBGLoadingKit_man.pdf brochure_liveblazer_FRETBGLoadingKit]
Aug. 28th 2008
Sept. 11th 2008
Attendees:
Sabine Jägle, Phillip, Norman Kilb, Christian Müller,
Wolfgang Schamel, Simone Weber
Themes:
1. CMV
Promotor
Sabine tried to amplificate the CMV Promotor (in the vector) ->
it didn’t work!
Improvements:
- Cut the CMV promoter out (of the vector) and then try again
to amplificate it.
- Prove, if we used the right primer.
- Turn the temperature down a bit.
- Use a positive control to be sure the polymerase and buffer
did work
2. Antibodies
Probably we could get some antibodies against NIP from Schamel and his
group, so we could proof if our origamis have the NIP. Normally we
could see the antibodies on the AFM.
3. FACS
We measured the calcium influx on the FACS.
- Unfortunately we didn´t see a calcium influx in
the cytoplasma membrane when we incubated the cells with our origami.
- We have to try to increase the concentration of our origami
- For the next measurement we also have to proof that TA/MgAc
itself has no influence on the calcium influx.
4. Fura-Loading
Because the Fura stain didn´t work when we tried to measure
the calcium influx at the microscope (ZBSA), we wanted to repeat the
staining to see what we should change.
o Again the staining didn´t work ->
We have to ask Nitschke about the conditions.
o We also have to order the Fura-2AM.
5. NIP binding to the
cells
We also repeated the binding measurement at the microscope.
- Like last time both the NIP origami and the control origami
(without NIP) bound to our cells :-(
- We should do the next measurement in Ringer-solution.
Therefore we have to test if the origamis are stable in the
Ringer-Solution with 12,5mM Mg/Ac!!!
- We should repeat this measurement with B-Cells (with a
“NIP-receptor”), to see if the origami (with and
without NIP) bind to those. Maybe some surface structures of the T
cells bind unspecific to our DNA.
-> We can get the B-cells(2558Lδm/mb-1) from Schamel!
Sept. 18th 2008
attendees
Philipp Mappes, Katja Arndt, Kristian Müller, Normann Kilb, Robert Gawlik, Simone Weber, Kathrin Pieper, Sabine Jägle
organisatory
- Bioss agrees to sponsorship
- team picture for wiki
- next meeting: wednesday, Sept. 24th 15.30
- lab: new box-arrangement (Normann)
- table for cloning strategy (Kathrin)
Ca-measurement
- date for measurement at the AFM (Nitschke)
- optionally: measurement at the Henneke lab --> children's clinic (Sabine)
- optionally: measurement at Imaging Facility in the medicin clinic --> searching for authorized contact-persons
Wiki
- carry over the inofficial wiki content to the official one (Robert und Philipp).
- final report about the whole project (everyone)
part-order
- ask for the ATG orders (need to send the plasmid again?) (Philipp)
- obscurities at order BCR-transmembraneregion --> control and correction(Kathrin)
- primer-order to synthesize signalpeptide by PCR (Kathrin)
control of DNA-Origami binding to cell-surface
- add wash-steps after addition of DNA-origami
- cells should keep in Mg2+ for measurement
- possibly the measurement can be carried out by ELISA --> immobilized AK to bind origamis; fluorophors fused to the oligos can be detected or use biotin-fused Oligos to get a detection by streptavidin fused enzyme reaction.
- control: monoclonal AK able to bind DNA-origami (Simone)
Sept. 24th. 2008
Sept. 29th 2008
Oct. 7th 2008'
Oct. 17th 2008
attendees:
Philipp Mappes, Katja Arndt, Kristian Müller, Normann Kilb, Robert Gawlik, Michael Kneib
Final report
- structure equal to a scientific paper containing the parts Abstract, Introduction, Materials and Methods, Results
- fusion of all sections we worked in: Origami, AFM, FACS, CA2+/Fura measuremt, cell cultures, modeling, ethics
- quotation of literature through autor and year of publication in the text (Max Muster, 2007), full quotation in references in the end
- deadline Friday Oct. 24th 2008
official wikipediasite
- continue the labjournal on the official page only
- not too much linking: maintenance of the flow of information
- all embedded files should start with "Freiburg2008"
iGEM parts
- uploading the parts starting on Monday Oct. 20th 2008
Oct. 21th 2008
attendees:
Moritz Busaker, Philipp Mappes, Kristian Müller, Normann Kilb, Robert Gawlik, Michael Kneib, Kathrin Pieper, Simone Weber
Cloning:
At least - All the constructes are cloned!!!
construcs:
pMA - singalpeptide – scFv-anti-NIP – gggs-Linker – transmembran region - X – pMA
X :
- bla1(1/2 beta-Lactamase)-
- bla2 (1/2 beta-Lactamase) –
- split-linker-C-CFP(Cerutan) –
- N-CFP –
- split-linker-C-GFP (split venus)–
- N-GFP –
- luciferase 1 (58) –
- luciferase 2 (57) –
Next steps:
1) We want to test, if the transmembran region really is located in the membrane
Therefore we want to clone a constructe which expresses a YFP on the cytoplasmic side of the receptor.
Construct:
- singalpeptide – scFv-anti-NIP – gggs-Linker – transmembran region - split-linker - bla1 - YFP -
2) We also want to test, if we able to bringe the receptors together by using a NIP coupled peptide.
Therefore we need the peptides from Schamels group -> Norman will ask them.
3) Origami-We want to try if the Origami are able to bring two or more receptors together.
Have to produce new Origami -> Michael and Norman
Next meeting: Oct. 23th 2008
|
 "
"

