Estimation of parameters
If we want to use the promoters used for the formation of the flagella (Description of the project), we will have to clearly defined their dynamics. To do so, a rather huge experimental work will be undertaken, consisting in providing the so-called 'Hill functions' for each promoters.
getting a Hill function from convenient datas
Therefore, we have written a little module which can estimate the parameters of the 'Hill functions', even with some noise and few data available.
Some details and the corresponding code can be found here : Programs.
The method we have employed is just based on a least-square optimization. Then, it could be generic enough for many applications and we would be glad to share the code if you feel it could be usefull.
getting convenient datas
Thus, we need experimental datas. To quantify the strength of a transcription factor on a promoter, we will use measurements of GFP fluorescence, and compare to the strength of the constitutive promoter [J23101], as it was proposed by the iGEM competition.
The datas we are looking for must appear as a table of values, giving several 'transduction rates' with their corresponding 'transcription factor concentrations'.
first hypothesis
For this aim, we made several hypothesis, which we will verify as good as it is possible for us :
(1) We do not take into acount the 'traduction' phase, so we directly correlate the transcription of a gene with the concentration of its protein.
(2) We assume that, whatever is the gene behind the promoter, its expression depends only of the transcription factor of the promoter, and not, for instance, of the weight of this gene. That's why comparing promoter strength is relevent only if the genes behind have similar length.
(3) We consider that the activity of a promoter is well described as a Hill function of its transcription factor (TF).
Thus, we suppose that the protein concentration (Prot) follows this equation :
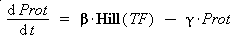
where gamma is a constant, due to degradation and of dilution of the protein, along time and cell divisions.
Therefore, if we consider a steady-state, for given concentration of the transcription factor, we will have :
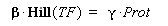
(4) Endly, knowing gamma will give us the kind of datas we are looking for. In a first approach, we assume that, as long as the barcteria are in their phase of exponential growth, the degradation is far smaller than the dilution, and can be omitted. But we will probably discuss that later.
(5) Unless we find further documents dealing with the relation between the intensity of fluorescence and the concentration of GFP, we will directly use the measure in fluorescence, that we will treat as a protein concentration, more or less arbitrary normalised.
how to control the concentration of the transcription factor ?
Now, we must use as a variable of reference an element that could be introduced in the bacteria, well-controlled, and from which will depends all the concentrations of our transcription factor. We propose a construction in which our transcription factor is put after the promoter Plac, which is under the repression of LacR. Since IPTG is a small diffusive molecule that binds to LacR and inhibits this way the repression of Plac, we can use it as an 'inducer'. To do so, we must place in the bacterium the gene lacR after a constitutive promoter (like J23101). According to previous hypothesis, this will provide at steady-state a 'constant concentration' of LacR (we note [LacR*], and it is supposed to be the TOTAL concentration of LacR, under every form) in the bacterium. If we consider the binding reaction this way (where LacR_IPTG denotes the complex)
LacR + IPTG ⇄ LacR_IPTG
with a dissociation constant K, we find at the steady-state
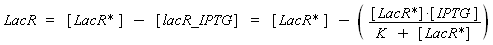
where [IPTG] denotes the concentration of IPTG we introduced in the medium, that will stay constant in all the bacteria along time, assuming that its degradation is near 0, and that the diffusion is quick. (We can already notice an other limitation in our protocol : this formula has got sense only for [IPTG] < (K + [LacR*]), that limits the range of [IPTG] we can use to determine the functions below. Notice that this limitation is conditionned by [LacR*], which depends of the constitutive promoter we put before lacR.)
According to the hypothesis (3), the activity of Plac would verify (keeping the same notations) :
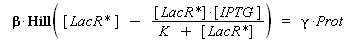
so, if k denotes the dissociation constant of (LacR + Plac ⇄ LacR_Plac),
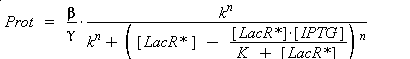
In the last equation, we will have 'access' (see hypothesis (5)
) to Prot and possibly to gamma, and we are looking for beta, k and n, thanks to our program (see getting Hill function with convenient datas). But we need to know ([LacR*] - [LacR*].[IPTG]/(K + [LacR*])), and we just have [IPTG]. However, we can reduce the last equation to
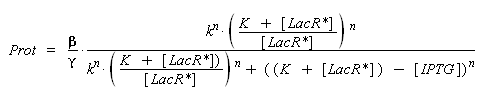
Thus, we have two possibilities :
- we can write a new algorithm that optimise an approaching solution of the new parameters, based on the same principal than 'findparam'.
- better but much longer and requiring much more precision, we can use the already noticed properties : [IPTG] < (K + [LacR*]). By having a look on the first equation of this section, we understand that beyond this limit, [LacR_IPTG] will no more evoluate. By observing the evolution of the influence of a growing (by steps as small as possible) concentration of [IPTG] introduced, we should be able to approximate the critic concentration when it no more changes, ~(K + [LacR*]). Less is the order n, better is the detection of this critic concentration, because of the greater derivative of the Hill function for small values of LacR. Therefore we should keep this estimation only if we find n ~ 1. Then, by considering k*((K + [LacR*])/[LacR*]) instead of k, we should easily determine all the parameters we need, only thanks to 'findparam'.
what are we looking for ?
The different functions we would like to determine are the followings. They are linked to a basic description of the 'experimental protocol' that will allow us to get the expected datas. We decide to let the original promoters in the bacteria, so that the strength that we are measuring is 'the strength for an additional promoter in the cell', keeping those who already exist.
According to the hypothesis (1) and (2), we assume this will directly give us [Protein] = ƒ1(IPTG), for a given Protein coded by a gene put behind the Plac promoter.
|
 "
"


