x
15th July
Conference in Edinburgh
Meet with the other UK iGEM teams.
16th July
Conference in Edinburgh
Presentation of projects to the other teams and to the attendees of the SynBioStandards network meeting.
17th July
Progress
-Ordered mutant E.coli chassis to test: Kch-, KdpD-, KdpE-, KdpF-E-
-Designed two plasmids:
-Sourced V.harveyi BB170 culture to obtain glutamate-gated potassium channel gene
-Modelled protein structure
-Designed primers for extraction of Kdp Operon from E.coli and Glu Gated channel gene from V.harveyi
-Meeting with Julia Davies
-Discussed feasibility
-Osmotic problems with ion sequestration, possible solution - "inert" sugars such as sorbitol and manitol
-Glutamate/glutamine specificity problems, may need glutamine- as well as glutamate-free growth medium
-Need to test efficacy of voltage creation over range of osmolarities, use flame photometer to plot total K+ as fn of growth curve
-Discussed Kch- mutants, generally benign
-Potential PD measurement methods: high resistance input voltmeter to minimise noise, glass capillary microelectrode, oxygen electrode
-Measurement issue of cells acting as charged particles, neutralising charge around electrode
-Decided on oxygen electrode for small voltage change measurements
-Extracted BioBricks from registry to use in plasmids:
- Terminators BBa_B1002 and BBa_B1006
18th July
Wet Work
There was no growth on the plates spread yesterday. There has been a few adjustments to the protocol.
Biobrick Extraction
| Class
| Registry
| Part Kit Location
| Tube Label
|
| Promoter | BBa_J23100 | 2008 1002-10E | F
|
| RBS | BBa_B0030 | 2008 1002-5G | G
|
| Stop | BBa_B1002 | 2008 1016-8A | H
|
| Stop | BBa_B1006 | 2008 1016-8D | I
|
| Promoter | BBa_J23113 | 2008 1002-12B | J
|
| Promoter | BBa_J23114 | 2008 1002-12C | K
|
| Control | pUC19 | | L
|
Bigger, better, faster, stronger
We found we could not extract enough DNA from the registry, so we are upgrading the extraction to the "bigger, better, faster, stronger" method.
Warm 50μL of EB in Eppendorf tubes at 50°C and add 4 punched spots. Keep it warming at 50°C for 20mins and spin down for 3 minutes at 15,000 g. Warm agin for 10mins and spin down again for 3mins. Pipette off the liquid which should have the DNA in.
We then confirmed with PCR.
Each sample except the control was then put through 34 cycles of PCR.
The following were added to eppendorf tubes:
5μL of DNA in EB buffer
2.5μL of each primer for Biobrick vectors
25μL of Finnzymes mastermix
15μL of sterile distilled water
And all 50μL were then run through the PCR reaction and 17μL of product run on an E-Gel with 3μL dye.
The results gave large amounts of DNA in each lane for all Biobricks except F.
The original extracts of G,H,I,J,K and L were then used for the following transformations.
To chilled tubes add 5μL of DNA + EB solution to 5μL of Cells ( Chemically competent Top 10 )
Ice for 30 minutes
Heat shock at 42°C for 60 seconds
Ice for 2 minutes
Add 500μL of SOC
Incubate at 37°C overnight
Prepare Agar plates : Add 200μL of Amp (100mg/mL) into 200mL of LA ( Final conc. of 100μg/mL ) and add 100μL of Amp (100mg/mL) into 200mL of LA ( Final conc. of 50μg/mL )
Plate neat samples of G,H,I,J,K,L onto both types of plate and incubate overnight.
21st July
Wet Work
Aims for Today
The results of yesterday's modified protocol still yielded no growth on the plates. Using yesterday's PCR product our aims were:
- Cut promoters with spe1
- Cut RBS with Xba1
- Ligate these together and perform further PCR amplification of linear fragments.
- Cut first 'stop' with spe1
- Cut second 'stop with xba1
- Ligate these together and perform further PCR amplification of linear fragments.
- Finally, put all the above elements into a vector plasmid.
- End of day: Aims completed
Length
- Promoter = 35 bases
- RBS = 16 bases
- Stop (B1002) = 35 bases and Stop (B1006) = 39 bases
22nd July
Wet Work
- Inoculated 10mL LB with E.coli strain MG1655
- Inoculated 10mL LB with E.coli strain TOP10 transformed with BioBrick J5311
16 LA plates were poured with 8 having 25μg/mL Kanamycin, ready for mutant strains to be grown on.
The gel extracts from the ligation of primer and rbs were extracted using Zymoclean gel extraction kit. Hopefully these now contain linear DNA of BBa_J23113 and BBa_B0030, and BBa_J23114 and BBa_B0030.
BioBricks J45996, J45995 and J45250 were also extracted from the registry, using the "bigger, better, faster, stronger, method", in order to provide template to extracted the OsmY promoter.
23rd July
Wet Work
- Recovered Biobrick plasmid YFP from previous culture.
- 1.6 ml of cells according to Zippy Plasmid Miniprep Kit protocol (60μL Elution Buffer used)
- Added 20ml LB to 10ml innoculation of E Coli MG1655 (see 22/07/08) to increase volume in preparation for experiments tomorrow on K+ tolerance in media.
24th July
Double Terminator
The double terminator was made from BBa_B1006 and BBa_B1002, combined together.
First the two Biobricks were cut using the fast digest enzymes, at either Xba or Spe. They were then run on an agarose gel, cut out and extracted, using Zymoclean gel extraction. The 2006 was not cut succesfully with Spe, so was not extracted. The samples were then ligated, using fast ligation, such that 1002-1002 and 1002-1006 were hopefully produced. This was done by combining the 1002 cut with Spe with the 1002 cut with Xba and the 1006 cut with Xba. These were then run on an agarose gel and cut out, then frozen overnight.
25th July
Double Terminator
The DNA was extracted from lanes 3&4 of the gel and extracted using the Zyppy Gel Extraction kit.
The extracted DNA was then PCRed using 34 cycles, and then 10μL was frozen at -20°C.
8μL of the PCR product was cut with both EcoR1 and Pst1, as was 5μL of the death plasmid, PSB4C5, using the fast digest protocol.
The gene was then ligated into the plasmid backbone, using the fast ligation kit, from Finnzymes.
500μL of normal TOP10 cells were made competent, and then transformed with the 4μL of the new plasmid, and grown on chloramphenicol overnight. 1μL of Puc19 was used as a control.
K+ assay for E.coli
Solutions of potassium chlotide were made up from 0mM to 500mM. 100μL of cells, 100μL of 10x KCl solution and 800μL of SDW were mixed and the OD600 measured every 30 mins, after agitation to ensure homogenaity. After 4 hours 800μL of the cell solution were spun down and resuspended in 1mL SDW, lysed using the freeze-thaw method, and the K+ concentration measured using the Flame Photometer.
28th July
The flame photometer was also calibrated using μM concentrations of potassium.
The mutants were streaked out from the innoculation discs onto both kanamycin and non antibiotic plates for the resistant strains, and just onto the normal plates for non-resistant strains. They were then incubated overnight at 37°C.
Mutant Plates
29th July
- Mutant E.coli strains had grown on plates.
- Transferred mutants to liquid culture.
- Primers arrived! Prepped as described in Protocols page
- PCR started with OsmY (from Biobrick plasmids) and KDP (from E. Coli MG1655 Cell Culture)
- E-Gel (SYBR) to confirm OsmY
- E-Gel (EtBr) to confirm Kdp and OsmY
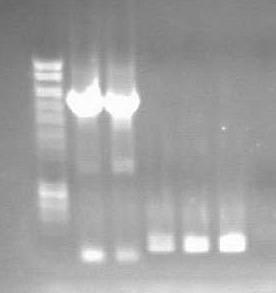
Gel showing KDP (2 thick bands on the left and OsmY (dimmer bands to the right)
30th July
Competence test
- Transformed PUK plasmid and YFP plasmid into mutant E.coli strains.
osmY and Kdp
- digested PCR products, and death gene vector with EcoR1 and Pst1
- ligated new BioBricks into vector
- Transformed into competent TOP10 cells
- Plate out onto Chloramphenicol plates
31st July
Mutants
- Mutant strains were grown in different potassium levels: 1mM, 50mM and 200mM.
- Growth curves were plotted, using OD600 readings every 30 minutes for 3 hours.
Kdp
- Ligated Kdp into death vector PSB4C5 again.
- More PCR of gene from MG1655.
OsmY
- TOP10 containing osmY-containing plasmid were grown in LB.
1st August
Mutants
- The OD600 of the mutants grown in different K+ concentrations was taken again.
osmY
- Plasmid mini prep was done.
- Gel was run, showing clear band at around 450bp, indicating osmY with BioBrick primer prefix and suffix.
- Glycerol stocks were made of the TOP10 cultures with the osmY-containing plasmid.
- More LB was added to the TOP10 cultures and they were put in the incubator.
Plasmid
- PSB4C5 was extracted from DB31, into 100μl SDW - final conc. 106.5ng/μl.
4th August
Biobricks
- The liagtion reaction from last week was drop dialised and then 5μL used to transform highly competent TOP10, before plating on chloramphenicol plates.
- The Biobrick combination of promoter-RBS were then cut with Pst1 and EcoR1 and ligated into pSB4C5 and then transformed into TOP10 and incubated overnight at room temperature in 600μL SOC.
5th August
Kdp?
- There were transformants from yesterday's transformation

James celebrates apparent success
- After Biobrick PCR the gel showed short fragments, suggesting self ligation of the vector.

Ellis realises the sad truth
- Repeated digest and ligation protocol from the weekend, with EcoR1 and Pst1 instead of Xba1 and Spe.
Biobricks
- Extracted two GFP-only sequences (J04630 and I714062) from the registry, for use as reporters for osmY.
- PCR on these and the RBS B0030. Ran E-gel to check PCR.
- Cut osmY plasmid with Spe, and RBS B0030 with Xba, and ligated the two.
- Transformed cells with this construct. Then realised this was silly.
6th August
Kdp?
- Transformants!
- But... PCR showed that none contained the Kdp insert.
- Gelled cut vector and extracted backbone band to use for ligation.
- Ligated Kdp gene into backbone and transformed cells. Again.
Mutants
- Grew up 5 mutants in LB, 1mM, 50mM and 200mM KCl.
7th August
Flame Photometry
- Various concentrations of KCl were made up in LB to calibrate for LB instead of water.
- Took OD600 readings of mutants grown in KCl concentrations in shaking incubator overnight
- They were then spun down, resuspended in SDW and spun again to wash, then resuspended.
- The resuspended cells were put through the flame photometer to discern internal cell K+ concentration.
- The supernatant K+ concentration was also measured.
Kdp
- Ran ligation mixtures on agarose gel and extracted 8kb (supercoiled plasmid) bands.
- Transformed cells with cleaned up plasmid.
Mutants
8th August
Kdp?
- Plates from yesterday showed no transformants. Others having difficulty with transformation too.
- Made up new Ampicillin stock and got new SOC and CaCl2 to try control (PUC) transformation in TOP10 next week.
Mutants
- Grew up 5 mutant strains in 5 different KCl concentrations in LB:
| Strain and Mutation
| KCl conc (mM)
| OD Reading
|
| BW25113
| 10
|
|
|
| 20
|
|
|
| 50
|
|
|
| 100
|
|
|
| 200
|
|
| BW30166
| 10
|
|
|
| 20
|
|
|
| 50
|
|
|
| 100
|
|
|
| 200
|
|
| JW0045
| 10
|
|
|
| 20
|
|
|
| 50
|
|
|
| 100
|
|
|
| 200
|
|
| JW1242
| 10
|
|
|
| 20
|
|
|
| 50
|
|
|
| 100
|
|
|
| 200
|
|
| JW3314
| 10
|
|
|
| 20
|
|
|
| 50
|
|
|
| 100
|
|
|
| 200
|
|
11th August
Kdp?
- PCR colonies on plates - no insert seen.
- Transform overweekend ligation.
OsmY and RBS
- PCR B0030 and restrict 10μL with Xba.
- Plasmid extract OsmY and restriction digest with Xba.
- Ligate and transform cells with 5μL
12th August
Kdp
- Again try ligation of Kdp and Vector backbone
OsmY
- Again try ligation of OsmY and B0030 backbone
- This time try using Xba for B0030 and ligate. Gel again and cut out linear fragment. Cut with Spe and gel - cut out circular fragment.
Make competent stocks
Chemically competent TOP10 cells were made by growing overnight in 200ml LB in a shaking incubater at 37°C.
200ml were spun down at 3800rpm for 8 mins, resuspended in 20ml CaCl2, spun down again and resuspended in 4ml CaCl2.
Again they were spun down (6500 rpm for 5 mins)and resuspended in 4ml 60%glycerol, spun down and resuspended in 2ml 60%glycerol. Finally they were aliquoted (100μL) into stock tubes and flash frozen in liquid nitrogen.
Electroporation competent TOP10 and DH5α cells were made by growing overnight in 200ml LB in a shaking incubater at 37°C.
100ml were spun down at 3800rpm for 8 mins, resuspended in 25ml SDW, spun down again and resuspended in 2ml SDW.
Again they were spun down (6500 rpm for 5 mins)and resuspended in 2ml 60%glycerol, spun down and resuspended in 2ml 60%glycerol. Finally they were aliquoted (100μL) into stock tubes and flash frozen in liquid nitrogen.
However lost half of the DH5α when centrifuge tube broke.
13th August
Kdp
Recovering frozen cells
To recover frozen cells they were thawed on ice and spun down at6500rpm for 5mins. They were then resuspended in 100μL CaCl2.
They were then transformed with 2μL pUC9 and grown on 100μg/mL Amp plates.
14th August
Kdp
- Cut Kdp with EcoR1 and Pst1 and ran on gel.
OsmY and RBS
- Cut with Spe and Xba respectively and ligated, using cooling ligation steps (42 - 35°C) and run on gel.
15th August
Kdp
- Nothing worked. Again. Query gremlins? Or possible sabotage by people in 106.
MEGA Mutant Assay
- THAT'S NUMBERWANG! (Actual Data)
16th August
Bank Holiday. Apparently no-one was quite that dedicated to come into work.
19th August
Ligation Diagnostic
- Due to lack of kdp insert following scPCR of transformed colonies, the ligation procedure was tested, involving Kdp & Vector biobricks together and separately, dirty ligations, negative controls where buffer was added but no ligase etc.
- Results of gel seemed inconclusive as hyperladder I ran very badly, but there seemed to be no DNA in the 'vector only lane' on both occasions.
- Conclusion: investigate our supply of pSB4C5 vector.
Kdp
- YPF biobrick plasmid used to provide vector backbone and a Kdp digest/ligation was attempted.
20th August
pSB4C5 & RBS
- scPCR perfomed on 5 colonies of DB31 & TOP10 to determine whether they contain pSB4C5 and B0030 respectively.
- Sucessful colonies grown up in LB, and pSB4C5 glycerol stocks were created.
- Large quantities of pSB4C5 were also harvested.
Kdp
- Electroporation of cells with the Kdp-YFP backbone.
- This failed (Even control, Jim!)
21st August
OsmY RBS
- B0030 harvested from LB stocks.
- Cut OsmY with Spe1 & B0030 with Xba1
- Attempted to ligate promoter and RBS sequences together again.
22nd August
Kdp & OsmY-RBS
- Digestion/ligation & drop dialysis prior to transformation into TOP10.
26th August
=Kdp screening
- Many transformant colonies were found on chloramphenicol plates.
- Single colony PCR was perfomed over a selection of 44 colonies using forward and reverse biobrick primers in order to gauge the size of the vector insert by gel visualisation.
- All screened colonies were negative for Kdp - concluded most likely to be blunt ended self ligation of pSB4C5 vector (without death gene).
27th August
OsmY RBS
- Primers containing RBS for osmY Biobrick were received
- PCR was performed from biobrick J45250
- Ligated into dephosphorylated vector backbone
- Colonies transformed
Restriction Digest & Ligation Test
- Various combinations of restriction digests & T4 phage genome ligation test.
- All results positive :-)
29th August
GluR0
- We received our synthesised plasmid from DNA 2.0 today!
- Transformed into TOP10.
File:Glur0.tif
2nd September
WE HAVE KDP!
- It has successfully been ligated into the death plasmid, transformed into TOP10 and plasmid prepped.
- The beautiful gel (note lanes 2-4 of right-hand gel, showing 7kb supercoiled plasmid):
5th September
STOP
- Transform B1002 and B1006 into TOP10
GluR0
- Cut with E+P or E+S
- Cut pSB4C5 with same
- Dephosphorylate pSB4C5
- Gel Extract, Ligate, Transform.
6th September
Ligations
- KDP and OsmY into pSB4C5 using E+S or X+P respectively
- Attempt to double stop with B1006 and B1002
9th September
Transformation
- Ligations from previous day transformed into TOP10
10th September
Analysis
Ligation
- Re-attempt KDP/OsmY ligation
11th September
Ligation
12th September
Analysis
- Analyse KDP, OsmY, Glur0 ligations
15th September
Plasmid Prep
Restriction Digest
- Digested prepped plasmids and run on gel.
16th September
Ligations
- GluR0 + pSB4T5 or pSB3T5
- B1002 + B1006
Gel
- B1002 + B1006 ligation product run with GluR0 unsupercoiled
- GluR0 was lightly irradiated to induce sing strand breaks to allow it to de-supercoil and act as a 4kb template.
18th September
Applications
- Designed standard infusion primers for biobrick assembly. Abandoned due to unfeasibly large scar sites between Biobricks, more effective to use custom designed primers for each reaction.
19th September
GluR0 Testing
- Tested GluR0 expression cells with shiny/expensive/confusing/very, very, very old electrical equipment. Engineers left. Damn (and blast) them. The filthy varmints.
- Initial results promising.
22nd September
PCR
- Single colony PCR of GluR0, OsmY, KDP.
- Results were ambiguous or negative.
23rd September
Plasmid Prep
- Minipreps of some cells from the previous day
Gel
- Plasmids from miniprep run on gel.
- Results negative or ambiguous.
24th September
Restriction Digest
- GluR0 plasmid cut (E or E+P) and run on gel.
- Results negative
Plasmid Prep
- KDP and some OsmY colonies. Run on gel. Best colony selected for continued work.
25th September
Plasmid Prep
- Miniprep on best cells containing KDP, OsmY and GluR0.
Restriction Digest
- Plasmids cut with various combinations of enzymes.
- Run on gel, results ambiguous.
26th September
Sequencing
- Samples PCRed and cleaned, prepared for sequencing.
x
 "
"







