|
|
| (58 intermediate revisions not shown) |
| Line 1: |
Line 1: |
| - | </style>
| |
| - | </html>
| |
| | {{:Team:Newcastle University/Header}} | | {{:Team:Newcastle University/Header}} |
| | {{:Team:Newcastle University/Template:UnderTheHome|page-title=[[Team:Newcastle University/Notebook|Wet Lab]]}} | | {{:Team:Newcastle University/Template:UnderTheHome|page-title=[[Team:Newcastle University/Notebook|Wet Lab]]}} |
| - | <div>
| + | {{:Team:Newcastle University/WetlabSideBar}} |
| - | {{:Team:Newcastle University/Template:PostItBox | + | |
| - | |boxtype=bluewhitebox
| + | |
| - | |title=Other progress
| + | |
| - | |detail-text=[[Team:Newcastle University/Meetings|Meetings]]<br>[[Team:Newcastle University/Protocols|Protocols]]<br>[[Team:Newcastle University/Results|Results]]
| + | |
| - | |link=}}
| + | |
| - | </div>
| + | |
| | <div id="maincol"> | | <div id="maincol"> |
| | | | |
| | + | ==Introduction== |
| | | | |
| | + | The overall design was too complicated for the time and resources available so we devised a proof of concept Brick which represented an input node from our full design. This Brick demonstrates that it is possible to transfer two-component peptide sensing from one strain into another and have it function as it did in the first strain. We took the subtilin two component system from ''Bacillus subtilis'' ATCC6633 and integrated it into the chromosome of ''B. subtilis'' 168, the typical laboratory ''B. subtilis'' strain at the ''amyE'' locus. |
| | | | |
| | + | The wet lab work involved: |
| | + | * Cloning the synthesized Brick from pUC57 in ''E. coli'' into our ''Bacillus'' integration vectors (with GFP and mCherry respectively downstream of the Brick) |
| | + | * Validation of the integration vector |
| | + | * Transformation and integration into the chromosome of ''B. subtilis'' 168 at the ''amyE'' locus |
| | + | * Validation of chromosomal integration |
| | + | * Characterisation of the integrated parts and their response to subtilin. |
| | | | |
| | | | |
| | + | ---- |
| | | | |
| | | | |
| - | | + | Our subtilin sensor, part [http://partsregistry.org/Part:BBa_K104001 BBa_k104001] |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | ==Introduction==
| + | |
| - | * We finally started in the labs today so we will start updating the wiki with what's been happening and the progress we are making.
| + | |
| | | | |
| | [[Image:biobrickk.gif|thumb|center|300px|Figure 1: Newcastle's iGEM team construct.]] | | [[Image:biobrickk.gif|thumb|center|300px|Figure 1: Newcastle's iGEM team construct.]] |
| | | | |
| - | Fig 1 shows the construct which contains:
| + | Figure 1 shows the construct which contains: |
| | * ''spaRK'' promotor | | * ''spaRK'' promotor |
| - | * ''rrnB'' - rRNA binding site
| + | * ''spaR'' (subtilin peptide antibiotic Regulation) - the 220 amino acid product of this gene usually regulates the downstream production of subtilin antibiotic. It has an N-terminal domain that can be phosphorylated and a C-terminal domian that has DNA binding properties [http://aem.asm.org/cgi/reprint/59/1/296.pdf] |
| - | * ''spaR'' (subtilin peptide antibiotic Regulation) - the 220 amino acid product of this gene usually regulates the downstream production of subtilin antibiotic. It has an N-terminal domain that can be phosphorylated and a C-terminal domian that has DNA binding properties [http://http://aem.asm.org/cgi/reprint/59/1/296.pdf] | + | * ''spaK'' (subtilin peptide antibiotic Kinase) - this gene codes for a 325 amino acid histadine kinase peptide that phosphorylates the N-terminus of ''spaR'' [http://aem.asm.org/cgi/reprint/59/1/296.pdf]. This activates the DNA binding ability of the C-terminus of ''spaR'', which in turn initiates transcription of the downstream gene. In the case of our construct, this gene is ''gfp''. |
| - | * ''spaK'' (subtilin peptide antibiotic Kinase) - this gene codes for a 325 amino acid histadine kinase peptide that phosphorylates the N-terminus of ''spaR'' [http://http://aem.asm.org/cgi/reprint/59/1/296.pdf]. This activates the DNA binding ability of the C-terminus of ''spaR'', which in turn initiates transcription of the downstream gene. In the case of our construct, this gene is ''gfp''. | + | * ''rrnO'' transcriptional terminator |
| - | * ''gfp'' (green fluorescence protein) - the marker being used to show activation of the ''spaRK'' system and therefore diagnosis of gram-positive bacteria by ''B. Subtilis'' | + | |
| | * ''spaS'' promotor - a strong promotor inducible by upstream activation of ''spaRK''. It can be placed in front any gene to regulate its activity. | | * ''spaS'' promotor - a strong promotor inducible by upstream activation of ''spaRK''. It can be placed in front any gene to regulate its activity. |
| | | | |
| - | '''Aim 1: clone the ''spaRK'' system from pUC57 into pJWV021 and transform into DH5 alpha competent ''E. coli'''''
| |
| | | | |
| | + | ---- |
| | | | |
| - | [[Image:cloning 1.jpg|thumb|center|300px|Aim 1]]
| |
| | | | |
| | + | '''Aim 1: clone the ''spaRK'' system from pUC57 into pJWV021 and transform into DH5 alpha competent ''E. coli''''' |
| | | | |
| - | '''Aim 2: clone the ''spaRK'' system from pUC57 into pGFP-rrnB and transform into TOP10 competent ''E. coli'''''
| |
| | | | |
| | + | [[Image:cloning 1.jpg|thumb|center|300px|Aim 1]] |
| | | | |
| - | [[Image:cloning 2.jpg|thumb|center|300px|Aim 2]]
| |
| | | | |
| - | * The 2.2kb fragment (ncl08) contains the ''spaRK'' system and promotor-less ''gfp'' linked to this. This means that when ''spaR'' is activated, its positive regulatory effect on spaK will in turn activate ''gfp''.
| + | ---- |
| | | | |
| - | '''Aim 3: clone the ''agr'' system from the plasmid vector into pGFP-rrnB and transform into DH5 alpha competent ''E. coli''''' | + | '''Aim 2: clone the ''spaRK'' system from pUC57 into pGFP-rrnB and transform into DH5 alpha competent ''E. coli''''' |
| | | | |
| | | | |
| - | [[Image:cloning 3.jpg|thumb|center|300px|Aim 3]] | + | [[Image:cloning 2.jpg|thumb|center|300px|Aim 2]] |
| | | | |
| - | * BBa_I647107 contains a partial ''agr'' operon, which includes ''agrC'' and ''A'' but has ''agrB'' and ''D'' deleted. This allows coding for the receptor (''C''/''A'') but not for production of the quorum peptides themselves (''B''/''D''). Eventually this will be linked to the ''spaRK'' system in a ''B. subtilis'' vector. | + | * The 2.2kb fragment (ncl08) contains the ''spaRK'' biobrick system. When properly cloned, our biobrick will replace the rrnB promoter in front of gfp present in plasmid pGFP-rrnB. Thus, gfp will now be under control of the subtilin inducible spaS promoter. This means that when ''SpaR'' is activated by SpaK (by sensing subtilin) its positive regulatory effect on PspaS will activate ''gfp''. |
| | | | |
| - | ==Monday 4th August==
| |
| | | | |
| - | * All 5 of us (Megan, Mark, Nina, Ria and Jess) went into the lab today to decide on a plan of action for the weeks to come.
| |
| | | | |
| - | * We used [[Agarose Gel Electrophoresis]] to check for the presence of plasmids (pGFPrrnB - our plasmid with the ''gfp'' gene, and pJWV021 - with the ''mCherry'' gene). Our gel was inconclusive, so we decided to re-run it the following day to confirm our results.
| + | ---- |
| | | | |
| - | * In the afternoon we attempted to transform TOP10 competent ''E. coli'' cells with DNA from the registry (from source plate 1010 well 4A and 1018 7A). See [[Transforming into DH5α or TOP10 E. coli]]
| |
| | | | |
| - | * We followed the extraction method outlined in the green folder and for each DNA spot we plated two agar dishes: one with a larger volume of DNA (4μl) and one with a smaller volume (2μl) to make colony counting easier. (See [[Making Agar Plates]].) We plated the following and incubated at 37˚C overnight:
| |
| | | | |
| - | '''Plate 1:''' +ve control (isolated plasmid plus TOP10 cells) | + | '''Aim 3: clone the ''agr'' (Cambridge biobrick for sensing S. aureus) system from the biobrick plasmid using EcoRI and SpeI sites into the corresponding sites of pGFP-rrnB. This will replace the PrrnB-gfp on the bacillus vector with the agr biobrick. The ligation was transformed into DH5 alpha competent ''E. coli''''' |
| | | | |
| - | '''Plate 2:''' -ve control (TOP10 cells only, no plasmid)
| |
| | | | |
| - | '''Plate 3:''' Large 1010 4A
| + | [[Image:cloning 3.jpg|thumb|center|300px|Aim 3]] |
| - | | + | |
| - | '''Plate 4:''' Small 1010 4A
| + | |
| - | | + | |
| - | '''Plate 5:''' Large 1018 7A
| + | |
| - | | + | |
| - | '''Plate 6:''' Small 1018 7A
| + | |
| - | | + | |
| - | ==Tuesday 5th August==
| + | |
| - | | + | |
| - | * Today we re-ran the [[gel]] of pGFP-rrnB and pJWV021 to check correct plasmid size and sufficient plasmid quantity. Results showed this to be true and the expected plasmid sizes of 8399bp for pGFP-rrnB and 7066bp for pJWV021.
| + | |
| - | | + | |
| - | ==Wednesday 6th August==
| + | |
| - | | + | |
| - | * Analyses of results from Monday’s transformation showed no colony growth except for the positive control. We decided to try another transformation into TOP10 E. coli using different spots from the registry. This time we used 1018 7A and 1004 6G.
| + | |
| - | | + | |
| - | * We noticed that Cambridge, who had also been having problems, had adapted the extraction method. So we tried their improved method, with a couple of minor differences:
| + | |
| - | | + | |
| - | '''1)''' Warm 50μl of H20 in an Eppendorf tube at 50˚C
| + | |
| - | | + | |
| - | '''2)''' Add 4 punched-out spots from the registry
| + | |
| - | | + | |
| - | '''3)''' Keep at 50˚C for 20 minutes
| + | |
| - | | + | |
| - | '''4)''' Centrifuge at 13,300g for 3 minutes
| + | |
| - | | + | |
| - | '''5)''' Warm for a further 10 minutes at 50˚C
| + | |
| - | | + | |
| - | '''6)''' Centrifuge at 13,300g for 3 minutes
| + | |
| - | | + | |
| - | '''7)''' Pipette out the supernatant which should (hopefully!) contain the DNA
| + | |
| - | | + | |
| - | | + | |
| - | * To see the original modified method, go to Cambridge’s OpenWetWare page:
| + | |
| - | http://openwetware.org/wiki/IGEM:Cambridge/2008/Protocols
| + | |
| - | | + | |
| - | ==Thursday 7th August==
| + | |
| - | | + | |
| - | * Unfortunately none of the plates showed any colonies. However, further incubation overnight of the plates at 37˚C from Monday 4th August produced 5 colonies on Plate 4 (2μl 1010 4A). These colonies were individually incubated overnight at 37˚C in 10μl LB. (See [[Making Overnight Cultures from Agar Colonies]].)
| + | |
| - | | + | |
| - | ==Friday 8th August==
| + | |
| - | | + | |
| - | * The 5 colonies from Thursday 7th August cultured in LB were pooled and the plasmid purified (see [[Isolating Plasmid from Cells (Miniprep)]]). When the sample was run on [[gel]] no band appeared, showing that the colonies were not ''E. coli'' and must have resulted from contamination.
| + | |
| - | | + | |
| - | ==Monday 11th August==
| + | |
| - | | + | |
| - | * Plasmids were [[isolated]] from pUc57-ncl08 (the ncl08 fragment contains the ''spaRK'' 2-part component system). We aim to clone this into pGFP-rrnB and pJWV021 vectors.
| + | |
| - | | + | |
| - | * Restrictions of pGFP-rrnB and pJWV021 carried out using 10μl plasmid and 0.5μl in 50μl total reaction volume (see [[Restricting Plasmids (Double Restriction)]]). pJWV021 was restricted in a 2-step reaction as we beleived that the enzymes were incompatible in the same buffer. The second restriction of this plasmid used 48μl of the purified plasmid mixture in a total volume of 100μl. pGFP-rrnB was restricted using EcoRI and NHeI. pJWV021 was restricted firstly with NHeI and secondly with BglII.
| + | |
| - | | + | |
| - | These were purified and stored overnight at -25˚C.
| + | |
| - | | + | |
| - | ==Tuesday 12th August==
| + | |
| - | | + | |
| - | * pGFPrrnB and pJWV021 were [[restricted]] using 10μl plasmid and 0.5μl each enzyme in 50μl total reaction volume:
| + | |
| - | - pGFPrrnB restricted with EcoRI + NheI
| + | |
| - | | + | |
| - | - pJWV021 restricted with NheI then BglII in a 2 step reaction (the second step used 48μl purified plasmid in 100μl total reaction volume).
| + | |
| - | | + | |
| - | Running the samples on a [[gel]] showed only partial restriction had occurred and that there was very little pJWV021.
| + | |
| - | | + | |
| - | ==Wednesday 13th August==
| + | |
| - | | + | |
| - | * pGFPrrnB and pJWV021 were [[restricted]] using 10μl plasmid and 0.5μl each enzyme in 50μl total reaction volume:
| + | |
| - | - pGFPrrnB restricted with EcoRI + NheI
| + | |
| - | | + | |
| - | - pJWV021 restricted with NheI then BglII in a 2 step reaction (the second step used 48μl purified plasmid in 100μl total reaction volume). These were stored overnight at -25˚C.
| + | |
| - | | + | |
| - | * BBa_I746101 (from Plate 1018 Spot 7A) and BBa_I746107 (containing the agr 2-component system) were received as cell stocks from the iGEM Registry.
| + | |
| - | | + | |
| - | ==Thursday 14th August==
| + | |
| - | | + | |
| - | * BBa_I746101 and BBa_746107 were [[isolated]] and run on a [[gel]] together with the 4 restrictions performed 13.08.08.
| + | |
| - | | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' empty
| + | |
| - | | + | |
| - | '''Lane 3:''' pGFPrrnB restricted with EcoRI + NheI
| + | |
| - | | + | |
| - | '''Lane 4:''' pJWV021 restricted with NheI + BglII
| + | |
| - | | + | |
| - | '''Lane 5:''' pUC57-ncl08 restricted with EcoRI + NheI
| + | |
| - | | + | |
| - | '''Lane 6:''' pUC57-ncl08 restricted with NheI + BglII
| + | |
| - | | + | |
| - | '''Lane 7:''' BBa_746101
| + | |
| - | | + | |
| - | '''Lane 8:''' BBa_746107
| + | |
| - | | + | |
| - | The gel showed that there was very little pGFPrrnB (restricted with EcoRI + NheI) and no pJWV021. It was concluded that the 2 step restriction and purification method reduced the plasmid yield significantly. As it was later discovered that this method was unnecessary (as NheI and BglII are compatible using buffer M), it was decided to perform the restriction as a single step in future.
| + | |
| - | | + | |
| - | The gel also showed successful restrictions of the pUC57-ncl08 plasmid and lots of the two BioBrick samples.
| + | |
| - | | + | |
| - | * The 2.2kb fragments from the pUC57-ncl08 restrictions were cut out of the gel using a UV light box and scalpel blade (see [[Cutting a Specific Band from Agarose Gel]]). The resulting gel slices were then purified to remove all gel and enzymes (see [[Purifying DNA from Gel Slices]]).
| + | |
| - | | + | |
| - | ==Friday 15th August==
| + | |
| - | | + | |
| - | pGFPrrnB, pJWV021 and BBa_I746107 were [[restricted]] using 20μl plasmid and 1μl each enzyme in 40μl total reaction volume:
| + | |
| - | - pGFPrrnB restricted with EcoRI + NheI
| + | |
| - | | + | |
| - | - pJWV021 restricted with NheI + BglII
| + | |
| - | | + | |
| - | - pGFPrrnB restricted with EcoRI + SpeI
| + | |
| - | | + | |
| - | - BBa_I746107 restricted with EcoRI + SpeI
| + | |
| - | | + | |
| - | These were incubated at 37˚C for 1 hour and run on a [[gel]]. The gel showed that pGFPrrnB and pJWV021 had restricted well with EcoRI + NheI and Nhe +BglII respectively, but pGFPrrnB and BBa_I746107 restricted with EcoRI + SpeI had only partially restricted.
| + | |
| - | | + | |
| - | ==Monday 18th August==
| + | |
| - | | + | |
| - | * The gel slices containing the 2.2kb ncl08 fragment were purified and a small amount run on a [[gel]] to check for correct restriction. Unfortunately it appeared that due to a pipetting error the solution was too dilute and thereofre not enough of the plasmid had been loaded on the gel to see.
| + | |
| - | | + | |
| - | ==Wednesday 20th August==
| + | |
| - | | + | |
| - | * The dilute 2.2kb fragment was concentrated by repeated rounds of heating and cooling. This was purified to see if any plasmid had been obtained. As the sample was in 500μl buffer, it was split into 5 x 100μl aliquots. Each was purified separatly and then the samples pooled afterwards to maximise the yield of DNA. This was then run on a [[gel]].
| + | |
| - | | + | |
| - | * The gel showed no pGFPrrnB (restricted with EcoRI and NheI) and only a small amount of pGFPrrnB (restricted with EcoRI). Therefore pGFPrrnB and pUC57-ncl08 were [[restricted]] using EcoRI and NheI (as this also showed as absent on the gel).
| + | |
| - | | + | |
| - | * Following these restrictions, the samples were run on [[gel]] which again showed them to be absent.
| + | |
| - | | + | |
| - | ==Thursday 21st August==
| + | |
| - | | + | |
| - | * pJWV021 and pUC57-ncl08 were restricted using a different method:
| + | |
| - | | + | |
| - | '''pJWV021 stock'''
| + | |
| - | * 48μl MillQ H2O
| + | |
| - | * 8μl buffer M
| + | |
| - | * 20μl pJWV021
| + | |
| - | | + | |
| - | | + | |
| - | '''- Tube 1''' = 19μl stock solution + 1μl NheI + 1μl BglII
| + | |
| - | | + | |
| - | '''- Tube 2''' = 19μl stock solution + 1μl NheI
| + | |
| - | | + | |
| - | '''- Tube 3''' = 19μl stock solution + 1μl BglII
| + | |
| - | | + | |
| - | '''- Tube 4''' = 19μl stock solution
| + | |
| - | | + | |
| - | | + | |
| - | pJWV021 and pUC57-ncl08 were restricted using a different method:
| + | |
| - | | + | |
| - | '''pJWV021 stock'''
| + | |
| - | * 48μl MillQ H2O
| + | |
| - | * 8μl buffer M
| + | |
| - | * 20μl pJWV021
| + | |
| - | | + | |
| - | | + | |
| - | '''- Tube 5''' = 19μl stock solution + 1μl NheI + 1μl BglII
| + | |
| - | | + | |
| - | '''- Tube 6''' = 19μl stock solution + 1μl NheI
| + | |
| - | | + | |
| - | '''- Tube 7''' = 19μl stock solution + 1μl BglII
| + | |
| - | | + | |
| - | '''- Tube 8''' = 19μl stock solution
| + | |
| - | | + | |
| - | ==Friday 22nd August==
| + | |
| - | | + | |
| - | * Stock pGFPrrnB from -80˚C was plated onto ampicillin, spectinomycin and normal [[agar]] (control) and incubated overnight at 37˚C.
| + | |
| - | | + | |
| - | * 2μl pUC57-ncl08 and pJWV021 were run on [[gel]] to check quantity. This showed large quantities of both plasmids and therefore pUC57-ncl08 was [[restricted]] with EcoRI + NHeI and NHeI + BglII. Controls (buffer A and buffer M without enzyme) were used for both restrictions.
| + | |
| - | | + | |
| - | ==Tuesday 26th August==
| + | |
| - | | + | |
| - | * Due to unforeseen circumstances, restriction mixtures were put in the fridge on Friday 22.08.08. Therefore the restriction was allowed to run for a further 1.5 hours before running on a gel.
| + | |
| - | | + | |
| - | [[Image:Jelly 26-08-081.jpg|thumb|right|300px|Gel showing our pUC57-ncl08 restrictions plus controls]] | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' empty
| + | |
| - | | + | |
| - | '''Lane 3:''' pUC57-ncl08 restricted with EcoRI + NHeI
| + | |
| - | | + | |
| - | '''Lane 4:''' pUC57-ncl08 restricted with NHeI and BglII
| + | |
| - | | + | |
| - | '''Lane 5:''' pUC57-ncl08 buffer A control
| + | |
| - | | + | |
| - | '''Lane 6:''' pUC57-ncl08 buffer M control
| + | |
| - | | + | |
| - | '''Lane 7:''' pure pJWV021
| + | |
| - | | + | |
| - | [[Image:DSC01658f.jpg|thumb|left|200px|pGFP-rrnB colonies on +Spec agar plate]]
| + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | Results from overnight (ON) agar cultures:
| + | |
| - | | + | |
| - | - pGFPrrnB (amp) - no colonies
| + | |
| - | | + | |
| - | - pGFPrrnB (spec) - colonies present
| + | |
| - | | + | |
| - | - pGFPrrnB (no antibiotic) - colonies present
| + | |
| - | | + | |
| - | * ON cultures were made:
| + | |
| - | | + | |
| - | - pGFP-rrnB grown on ampicillin
| + | |
| - | | + | |
| - | - pJWV021 grown on ampicillin
| + | |
| - | | + | |
| - | - pUC57-ncl08 grown on ampicillin
| + | |
| - | | + | |
| - | ==Wednesday 27th August==
| + | |
| - | | + | |
| - | * ON cultures were analysed:
| + | |
| - | | + | |
| - | - pGFP-rrnB grown on ampicillin - cells had lysed (medium was clear with large bubbles at the surface).
| + | |
| - | | + | |
| - | - pJWV021 grown on ampicillin - frozen glycerol stock made (see protocol).
| + | |
| - | | + | |
| - | - pUC57-ncl08 grown on ampicillin - frozen glycerol stock made.
| + | |
| - | | + | |
| - | * A [[colony stab]] was taken of pGFP-rrnB using 10μl spectinomycin in 10mL LB as the culture medium.
| + | |
| - | | + | |
| - | ==Thursday 28th August==
| + | |
| - | | + | |
| - | * New [[frozen glycerol stock]] of cells made to be kept at -80˚C.
| + | |
| - | | + | |
| - | * [[Isolated plasmid]] from overnight culture of pGFP-rrnB. For this the 10mL culture was divided into 5 x 2mL plastic tubes and pelleted by centrifugation for 10 minutes. The supernatent was discarded and follwing the isolation procedure the product was pooled.
| + | |
| - | | + | |
| - | * [[Restrictions]] of pUC57-ncl08 and pJWV021 were carried out.
| + | |
| - | | + | |
| - | [[Image:Jelly 28-08-082.jpg|thumb|right|300px| Gel showing pUC57-ncl08 and pJWV021 restrictions.]]
| + | |
| - | | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' pUC57-ncl08 restricted with EcoRI
| + | |
| - | | + | |
| - | '''Lane 3:''' pUC57-ncl08 restricted with BglII
| + | |
| - | | + | |
| - | '''Lane 4:''' pUC57-ncl08 restricted with EcoRI and NHeI
| + | |
| - | | + | |
| - | '''Lane 5:''' pUC57-ncl08 restricted with NHeI and BglII
| + | |
| - | | + | |
| - | '''Lane 6:''' pUC57-ncl08 control (no enzyme)
| + | |
| - | | + | |
| - | '''Lane 7:''' pJWV021 restricted with NHeI and BglII
| + | |
| - | | + | |
| - | '''Lane 8:''' pure pGFPrrnB
| + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | * This gel shows that only partial restrictions of the plasmids had occurred. It was subsequently discovered that an incorrect quantity of buffer was being used. This was rectified in further restriction reactions.
| + | |
| - | | + | |
| - | ==Friday 29th August==
| + | |
| - | | + | |
| - | * Today we [[restricted]] our pUC57-ncl08 plasmid. In one reaction we used EcoR1 and Nhe1, and in other we used BglII and Nhe1. We also carried out a restriction with just BglII to check for the correct sized linear DNA. In addition we ran unrestricted plasmid on the gel.
| + | |
| - | | + | |
| - | '''Expected band sizes:'''
| + | |
| - | * For digestion with BglII only: ~ 4.9kb
| + | |
| - | * For digestion with Nhe1 and EcoR1: ~ 2.2kb and 2.7kb
| + | |
| - | * For digestion with Nhe1 and BglII: ~ 2.2kb and 2.7kb
| + | |
| - | | + | |
| - | [[Image:gelFINAL.jpg|thumb|center|300px|Gel showing successful digestions of pUC57-ncl08]]
| + | |
| - |
| + | |
| - | * As you can see, the restrictions appear successful.
| + | |
| - | | + | |
| - | ==Monday 01st September==
| + | |
| - | | + | |
| - | * To obtain a large enough volume for our ligations, we carried out all 4 [[restrictions]] again (i.e. of our pUC57-ncl08, pGFP-rrnB and pJWV021). We used a total volume of 100μl per reaction (using 2μl of each enzyme and 20μl of plasmid). These were incubated for 1 1/2 hours at 37˚C.
| + | |
| - | | + | |
| - | * 4μl of each reaction was then run on a [[gel]] for 1 hour @ 70V- See the gel below (Tuesday the 2nd)
| + | |
| - | [[Image:01091.jpg|thumb|right|300px|Gel showing pUC57-ncl08, pGFp-rrnB and pJWV021 restrictions ran on a gel after 1 hour 30 mins and 2 hours 30 mins incubation.]]
| + | |
| - | | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' pUC57-ncl08 restricted with EcoR1 and Nhe1 (1 hour 30 mins)
| + | |
| - | | + | |
| - | '''Lane 3:''' pUC57-ncl08 restricted with BglII and Nhe1(1 hour 30 mins)
| + | |
| - | | + | |
| - | '''Lane 4:''' pGFP-rrnB restricted with EcoR1 and Nhe1(1 hour 30 mins)
| + | |
| - | | + | |
| - | '''Lane 5:''' pJWV021 restriced with BglII and NHe1(1 hour 30 mins)
| + | |
| - | | + | |
| - | '''Lane 6:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 7:''' pUC57-ncl08 restricted with EcoR1 and Nhe1 (2 hours 30 mins)
| + | |
| - | | + | |
| - | '''Lane 8:''' pUC57-ncl08 restricted with BglII and Nhe1(2 hours 30 mins)
| + | |
| - |
| + | |
| - | '''Lane 9:''' pGFP-rrnB restricted with EcoR1 and Nhe1(2 hours 30 mins)
| + | |
| - | | + | |
| - | '''Lane 10:''' pJWV021 restriced with BglII and NHe1(2 hours 30 mins)
| + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | '''Expected band sizes:'''
| + | |
| - | * '''Lane 2:''' ~ 2.2kb and 2.7kb
| + | |
| - | * '''Lane 3:''' ~ 2.2kb and 2.7kb
| + | |
| - | * '''Lane 4:''' ~ 7.9kb and 0.5kb
| + | |
| - | * '''Lane 5:''' ~ 6.6kb and 0.5kb
| + | |
| - | | + | |
| - | * The gel shows partial restrictions in lanes 2 and 3. In Lane 4, the larger fragment is the correct size, but the observed smaller fragment is larger than expected. Lane 5 shows that no pJWV021 is present, so we will need to re-isolate this tomorrow.
| + | |
| - | | + | |
| - | * The samples were incubated for a further 1 hour, and run on a gel again. Unfortunately, there was no change, so we decided to re-isolate all 3 plasmids tomorrow.
| + | |
| - | | + | |
| - | * We made ON cultures using 10μl ampicillin in 10ml of LB for pUC57-ncl08 and pJWV021, and 5μl of spectinomycin in 10ml LB for pGFP-rrnB. These were incubated overnight at 37˚C.
| + | |
| - | | + | |
| - | ==Tuesday 2nd September==
| + | |
| - | | + | |
| - | * The 3 ON cultures were [[isolated]] by dividing each 20ml tube into 2 x 5ml tubes. These were centrifuged for 10 minutes to pellet.
| + | |
| - | | + | |
| - | * Once isolated, we ran 5μl of each sample on a [[gel]] to check for the presence of plasmid. As you can see from lanes 1-7 on the gel below, there was plenty of pGFP-rrnB but not much of pJWV021. Also, the pUC57-ncl08 samples look 'dirty' (see grey smears).
| + | |
| - | | + | |
| - | * As a result, for the digests we decided to use more of the pUC57-ncl08 and pJWV021 (in a total volume of 100µl for the digestions, we used 40µl of plasmid for the pUC57-ncl08 digestion, 30µl for the pJWV021 digestion and 20µl for the pGFP-rrnB digestion).
| + | |
| - | | + | |
| - | * After incubation, we ran the digest products on a [[gel]].
| + | |
| - | | + | |
| - | '''Expected band sizes:'''
| + | |
| - | * '''Lane 9:''' ~ 7.9kb and 0.5kb
| + | |
| - | * '''Lane 10:''' ~ 6.6kb and 0.5kb
| + | |
| - | * '''Lane 11:''' ~ 2.2kb and 2.7kb
| + | |
| - | * '''lane 12:''' ~ 2.2kb and 2.7kb
| + | |
| - | | + | |
| - | [[Image:02091.jpg|thumb|right|300px|Gel showing our 3 unrestricted plasmids, along with the 4 restrictions]]
| + | |
| - | | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' Undigested pGFP-rrnB sample 1
| + | |
| - | | + | |
| - | '''Lane 3:''' ‘’ ‘’ sample 2
| + | |
| - | | + | |
| - | '''Lane 4:''' Undigested pJWV021 sample 1
| + | |
| - | | + | |
| - | '''Lane 5:''' ‘’ ‘’ sample 2
| + | |
| - | | + | |
| - | '''Lane 6:''' Undigested pUC57-ncl08 sample 1
| + | |
| - | | + | |
| - | '''Lane 7:''' ‘’ ‘’ sample 2
| + | |
| - | | + | |
| - | '''Lane 8:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 9:''' pGFP-rrnB digested with EcoR1 and Nhe1
| + | |
| - | | + | |
| - | '''Lane 10:''' pJWV021 digested with BglII and Nhe1
| + | |
| - | | + | |
| - | '''Lane 11:''' pUC57-ncl08 digested with EcoR1 and Nhe1
| + | |
| - | | + | |
| - | '''Lane 12:''' pUC57-ncl08 digested with BglII and Nhe1
| + | |
| - | | + | |
| - | The gel showed that our restrictions were successful. Before we moved on to ligating, we purified the 4 restricted samples using the PCR DNA purification kit to remove the enzymes.
| + | |
| - | | + | |
| - | For the ligations we made each reaction mixture to 25µl (using 1.5µl of T4 ligase and 2.5µl of 10x ligase buffer)- see protocols tab for method. We incubated our samples for 1 hour in a 27˚C water bath.
| + | |
| - | | + | |
| - | ==Wednesday 3rd September==
| + | |
| - | [[Transformations]]
| + | |
| - | | + | |
| - | * Today our aim was to transform our ligated plasmids into DH5-aplha E.coli competent cells.
| + | |
| - | | + | |
| - | * For each transformation, we added 100µl of DH5-alpha E.coli competent cells to 12.5µl of the ligation mixtures from yesterday.
| + | |
| - | | + | |
| - | * So in total we carried out 6 transformations, preparing them all on ice:
| + | |
| - | | + | |
| - | '''T1)''' Transforming pGFP-rrnB containing the 2.2kb fragment from pUC57-ncl08 into DH5-alpha
| + | |
| - | | + | |
| - | '''T2)''' Transforming pGFP-rrnB minus the 2.2kb fragment from pUC57-ncl08 into DH5-alpha
| + | |
| - | | + | |
| - | '''T3)''' Transforming pJWV021 containing the 2.2kb fragment from pUC57-ncl08 into DH5-alpha
| + | |
| - | | + | |
| - | '''T4)''' Transforming pJWV021 minus the 2.2kb fragment from pUC57-ncl08 into DH5-alpha
| + | |
| - | | + | |
| - | '''T5)''' Positive control: DH5-alpha cells plus pGFP-rrnB
| + | |
| - | | + | |
| - | '''T6)''' Negative control: DH5-alpha cells only
| + | |
| - | | + | |
| - | * See protocols for method.
| + | |
| - | | + | |
| - | * The transformations were then plated onto antibiotic agar:
| + | |
| - | | + | |
| - | '''T1 :''' +spec
| + | |
| - | | + | |
| - | '''T2 :''' +spec (small and large volume plated)
| + | |
| - | | + | |
| - | '''T3 :''' +amp and +kan
| + | |
| - | | + | |
| - | '''T4 :''' +amp +kan
| + | |
| - | | + | |
| - | '''T5 :''' +spec
| + | |
| - | | + | |
| - | '''T6 :''' +spec
| + | |
| - | | + | |
| - | ==Thursday 04th September==
| + | |
| - | [[Image:plates1.jpg|thumb|right|300px|Our plates from Wednesday the 3rd]]
| + | |
| - | * The plates from 03.09.08 showed expected colony growth and therefore stab cultures were taken from individual colonies on Plates 1, 2 and 9. The culture media were made using 10mL of LB in 15mL plastic tubes together with the correct volume of the relavent antibiotic. This was 5μl spectinomycin for the GFP-rrnB colonies and 4μl kanomycin for the pJWV021 colonies. Cultures were taken as follows:
| + | |
| - | | + | |
| - | '''GFP-rrnB - pUC57 ligation (grown on spec)'''
| + | |
| - | * 9 white colonies (1-9) (Plate 1)
| + | |
| - | * 1 green colony (10) (Plate 1)
| + | |
| - | * 2 white colonies (11,12) (Plate 2)
| + | |
| - | | + | |
| - | '''pJWV021 - pUC57 ligation (grown on kan)'''
| + | |
| - | * 10 white colonies (13-22) (Plate 9)
| + | |
| - | * 1 pink colony (23) (Plate 9)
| + | |
| - | * 1 bright white colony (24) (Plate 9) (this is likely to a be contaminant non-E.coli colony)
| + | |
| - | | + | |
| - | * All 24 culture tubes were incubated at 37˚C whilst shaking for ~20 hours. This large quantity of cultures should ensure that we obtain at least 2 cultures (one GFP-rrnB and one pJWV021) that have taken up the ncl08 insert. The plates were kept at room temperature in case fulrther stab cultures needed to be made.
| + | |
| - | | + | |
| - | ==Friday 05th September==
| + | |
| - | | + | |
| - | * 4μl of each of the 24 overnight cultures was centrifuged and the plasmid isolated from the resulting pellets. The isolated plasmid was then restricted to see if the plasmid had taken up the insert. All pGFPrrnB-nvl08 plasmids were restricted using EcoRI and NheI and pJWV021-ncl08 plasmids restricted with NheI and BglII. The restrictions were carried out in using 7.5μl plasmid in a total volume of 15μl. Samples were run on gel
| + | |
| - | | + | |
| - | [[Image:Gel_2.jpg|thumb|right|300px|Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies]]
| + | |
| - | | + | |
| - | | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' pGFPrrnB-ncl08 colony 1 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 3:''' pGFPrrnB-ncl08 colony 2 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 4:''' pGFPrrnB-ncl08 colony 3 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 5:''' pGFPrrnB-ncl08 colony 4 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 6:''' pGFPrrnB-ncl08 colony 5 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 7:''' pGFPrrnB-ncl08 colony 6 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 8:''' pGFPrrnB-ncl08 colony 7 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 9:''' pGFPrrnB-ncl08 colony 8 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 10:''' pGFPrrnB-ncl08 colony 9 (white colony, 25μl plate)
| + | |
| - | | + | |
| - | '''Lane 11:''' pGFPrrnB-ncl08 colony 10 (white colony, 250μl plate)
| + | |
| - | | + | |
| - | '''Lane 12:''' pGFPrrnB-ncl08 colony 11 (white colony, 250μl plate)
| + | |
| - | | + | |
| - | '''Lane 13:''' pGFPrrnB-ncl08 colony 12 (green colony, 25μl plate)
| + | |
| - | | + | |
| - | [[Image:Jelly Cherry 05-09-082.jpg|thumb|right|300px|Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies.]]
| + | |
| - | | + | |
| - | | + | |
| - | '''Lane 1:''' 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2:''' pJWV021-ncl08 colony 1 (white colony)
| + | |
| - | | + | |
| - | '''Lane 3:''' pJWV021-ncl08 colony 2 (white colony)
| + | |
| - | | + | |
| - | '''Lane 4:''' pJWV021-ncl08 colony 3 (white colony)
| + | |
| - | | + | |
| - | '''Lane 5:''' pJWV021-ncl08 colony 4 (white colony)
| + | |
| - | | + | |
| - | '''Lane 6:''' pJWV021-ncl08 colony 5 (white colony)
| + | |
| - | | + | |
| - | '''Lane 7:''' pJWV021-ncl08 colony 6 (white colony)
| + | |
| - | | + | |
| - | '''Lane 8:''' pJWV021-ncl08 colony 7 (white colony)
| + | |
| - | | + | |
| - | '''Lane 9:''' pJWV021-ncl08 colony 8 (white colony)
| + | |
| - | | + | |
| - | '''Lane 10:''' pJWV021-ncl08 colony 9 (white colony)
| + | |
| - | | + | |
| - | '''Lane 11:''' pJWV021-ncl08 colony 10 (white colony)
| + | |
| - | | + | |
| - | '''Lane 12:''' pJWV021-ncl08 colony 11 (white colony)
| + | |
| - | | + | |
| - | '''Lane 13:''' pJWV021-ncl08 colony 12 (white colony)
| + | |
| - | | + | |
| - | | + | |
| - | * The gels showed correct insert fragments (2.2kb) in lanes 8 (colony 7) and 12 (colony 11) for pGFPrrnB and lanes 2, 3, 4, 5, 6, 7, 9 and 10 (colonies 13, 14, 15, 16, 17, 18, 20 and 21 respectively) for pJWV021.
| + | |
| - | | + | |
| - | * Undigested plasmid from colonies 7 + 11 (pGFPrrnB) and 13 + 14 (pJWV021) were transformed into ''Bacillus subtilis'' (see protocol section).
| + | |
| - | | + | |
| - | * Plasmid renamed once transformed into ''B. subtilis'' to avoid confusion with plasmid in ''E. coli'':
| + | |
| - | | + | |
| - | - pGFPrrnB-ncl08 (''E. coli'') = iGEMgfp (''B. subtilis'')
| + | |
| - | | + | |
| - | - pJWV021-ncl08 (''E. coli'') = iGEMcherry (''B. subtilis'')
| + | |
| - | | + | |
| - | * Agar cultures were also made from the same colony cultures:
| + | |
| - | | + | |
| - | - iGEMgfp (chloramphenicol) colony 7
| + | |
| - | | + | |
| - | - iGEMgfp (chloramphenicol) colony 11
| + | |
| - | | + | |
| - | - iGEMgfp (chloramphenicol) negative control
| + | |
| - | | + | |
| - | - iGEMcherry (kanomycin) colony 13
| + | |
| - | | + | |
| - | - iGEMcherry (kanomycin) colony 14
| + | |
| - | | + | |
| - | - iGEMcherry (kanomycin) negative control
| + | |
| - | | + | |
| - | ==Monday 08th September==
| + | |
| - | | + | |
| - | * Results from the agar cultures:
| + | |
| - | | + | |
| - | - iGEMgfp (chloramphenicol) colony 7 - many colonies
| + | |
| - | | + | |
| - | - iGEMgfp (chloramphenicol) colony 11 - many colonies
| + | |
| - | | + | |
| - | - iGEMgfp (chloramphenicol) negative control - no colonies
| + | |
| - | | + | |
| - | - iGEMcherry (kanomycin) colony 13 - a few colonies
| + | |
| - | | + | |
| - | - iGEMcherry (kanomycin) colony 14 - 3 colonies (plus 'background' contaminants)
| + | |
| - | | + | |
| - | - iGEMcherry (kanomycin) negative control - no colonies (some 'background' contaminants)
| + | |
| - | | + | |
| - | * Agar stabs were taken from the following colonies and streaked onto agar to obtain isolated colonies:
| + | |
| - | | + | |
| - | - iGEMgfp colonies 7.1, 7.2, 7.3, 7.4, 7.5, 7.6
| + | |
| - | | + | |
| - | - iGEMgfp colonies 11.1, 11.2, 11.3, 11.4, 11.5, 11.6
| + | |
| - | | + | |
| - | - iGEMcherry colonies 13.1, 13.2, 13.3
| + | |
| - | | + | |
| - | ''B. subtilis'' wild type strain ATCC6633 was also streaked onto agar.
| + | |
| - | | + | |
| - | * Overnight (ON) cultures were made by JW of iGEMgfp 7.1, iGEMgfp colonies 11.1, iGEMcherry 13.1 and 13.2.
| + | |
| - | The following cultures were then made and incubated at 37˚C whilst shaking for 3 hours. Dilute (dil) cultures were made by adding 500μl ON culture to 10mL LB. After 3 hours ATCC6633 supernatent was obtained by centrifuging ON ATCC6633 culture and 250μl of this added to the + ATCC6633 tubes.
| + | |
| - | | + | |
| - | - iGEMgfp ON + ATCC6633
| + | |
| - | | + | |
| - | - iGEMgfp ON - ATCC6633
| + | |
| - | | + | |
| - | - iGEMgfp dil + ATCC6633
| + | |
| - | | + | |
| - | - iGEMgfp dil - ATCC6633
| + | |
| - | | + | |
| - | - iGEMcherry ON + ATCC6633
| + | |
| - | | + | |
| - | - iGEMcherry ON - ATCC6633
| + | |
| - | | + | |
| - | - iGEMcherry dil + ATCC6633
| + | |
| - | | + | |
| - | - iGEMcherry dil - ATCC6633
| + | |
| - | | + | |
| - | * These were left to incubate at 37˚C for a further 2 hours to allow ''gfp'' and ''mCherry'' induction by the subtilin contained in the ATCC6633 supernatent. Those wiothout supernatent were positive controls.
| + | |
| - | | + | |
| - | * The ''B. subtilis'' cells were prepared for microscopy (see protocol) and viewed:
| + | |
| - | | + | |
| - | - iGEMgfp ON + ATCC6633 - some cells expressed ''gfp''
| + | |
| - | | + | |
| - | - iGEMgfp ON - ATCC6633 - some cells expressed ''gfp'' (although less than in the presence of subtilin)
| + | |
| - | | + | |
| - | - iGEMgfp dil + ATCC6633 - some cells expressed ''gfp''
| + | |
| - | | + | |
| - | - iGEMgfp dil - ATCC6633 - some cells expressed ''gfp'' (although less than in the presence of subtilin)
| + | |
| - | | + | |
| - | - iGEMcherry ON + ATCC6633 - very few celled expressed mCherry
| + | |
| - | | + | |
| - | - iGEMcherry ON - ATCC6633 - very few celled expressed mCherry
| + | |
| - | | + | |
| - | - iGEMcherry dil + ATCC6633 - very few celled expressed mCherry
| + | |
| - | | + | |
| - | - iGEMcherry dil - ATCC6633 - very few celled expressed mCherry
| + | |
| - | | + | |
| - | * There appeared to be a difference in the GFP intensities between iGEMgfp with and without subtilin but this microspy was not sufficient to confirm this.
| + | |
| - | | + | |
| - | * Starch, sucrose and glucose agar plates were made (see protocol) and the following colonies streaked:
| + | |
| - | - iGEMgfp 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 11.1, 11.2, 11.3, 11.4, 11.5, 11.6 streaked onto starch agar
| + | |
| - | | + | |
| - | - iGEMcherry 13.1, 13.2, 13.3 streaked onto both glucose and sucrose
| + | |
| - | | + | |
| - | * iGEMgfp should be able to grow on starch but will have no 'halo' surrounding the colonies (the halo is digested starch). This is becuase the insert should have integrated into ''amyE'' and the bacteria therefore cannot make amylase to digest starch.
| + | |
| - | iGEMcherry should not be able to grow on sucrose as the insert should have integrated into ''sacA''. The bacterium therefore cannot make sucrase to digest sucrose and consequently will have no carbon source. The glucose plates act as controls.
| + | |
| - | | + | |
| - | * Overnight cultures were also made og iGEMgfp 7.1 and 11.1 and iGEMcherry 13.1 and 13.2.
| + | |
| - | | + | |
| - | ==Tuesday 9th September==
| + | |
| - | | + | |
| - | * Half of the overnight cultures iGEMgfp 7.1 and iGEMcherry 13.1 were diluted (by adding 500μl ON culture to 10mL LB) to make the following cultures. These were incubated at 37˚C whilst shaking for 3 hours. 250μl ATCC6633 supernatent was then added to the + ATCC6633 tubes and the mixtures left to incubate for 2 hours at 37˚C.
| + | |
| - | | + | |
| - | * Glycerol stocks made (see protocol) of iGEMgfp 7.1 and 11.1 and iGEMcherry 13.1 and 13.2.
| + | |
| - | | + | |
| - | * Microscopy of the cultures showed similar results to yesterday with again only a possible difference between the iGEMgfp with and without subtilin.
| + | |
| - | | + | |
| - | * BBa_I746107 and pGFPrrnB restricted using 20μl plasmid and 2μl of each enzyme (EcoRI and SpeI) in 100μl total reaction volume. These were run on a gel at 70V for 1 hour.
| + | |
| - | | + | |
| - | '''Lane 1''' = 1kb ladder
| + | |
| - | | + | |
| - | '''Lane 2''' = pGFP restricted with EcoRI and SpeI
| + | |
| - | | + | |
| - | '''Lane 3''' = BBa_I746107 restricted with EcoRI and SpeI
| + | |
| - | | + | |
| - | * This showed successful restriction of both plasmids which were then purified and ligated overnight using 6μl plasmid and 1.5μl enzyme (T4 ligase) in 25μl total reaction volume.
| + | |
| - | | + | |
| - | ==Wednesday 10th September==
| + | |
| - | | + | |
| - | * Overnight ligation mixtures transformed into competent DH5α ''E. coli'' cells (see protocol).
| + | |
| - | - 12.5μl pGFPrrnB-BBa_746107 + 100μl DH5α cells
| + | |
| - | - 12.5μl pGFPrrnB + 100μl DH5α cells (positive control)
| + | |
| - | - 100μl DH5α cells (negative control)
| + | |
| - | | + | |
| - | * All transformants were then plated onto spectinomycin agar in both 25μl and 250μl volumes and incubated overnight at 37˚C.
| + | |
| - | | + | |
| - | ==Thurday 11th September==
| + | |
| - | | + | |
| - | * Single colonies of iGEMcherry were streaked from the overnight kanamycin plates onto sucrose and glucose plates.
| + | |
| - | | + | |
| - | * Stab cultures were taken from the overnight 25μl and 250μl pGFP-BBa_I7045898 plates (grown on spectinomycin) and grown in 10mL LB:
| + | |
| - | | + | |
| - | - 25μl plate white colony 1
| + | |
| - | | + | |
| - | - 25μl plate white colony 2
| + | |
| - | | + | |
| - | - 25μl plate white colony 3
| + | |
| - | | + | |
| - | - 25μl plate white colony 4
| + | |
| - | | + | |
| - | - 25μl plate white colony 5
| + | |
| - | | + | |
| - | - 25μl plate white colony 6
| + | |
| - | | + | |
| - | - 250μl plate white colony 1
| + | |
| - | | + | |
| - | - 250μl plate white colony 2
| + | |
| - | | + | |
| - | - 250μl plate white colony 3
| + | |
| - | | + | |
| - | - 250μl plate white colony 4
| + | |
| - | | + | |
| - | - 250μl plate white colony 5
| + | |
| - | | + | |
| - | - 250μl plate green colony 6
| + | |
| - | | + | |
| - | These were incubated overnight at 37˚C whilst shaking.
| + | |
| - | | + | |
| - | * iGEMgfp was diluted by adding 250μl of the overnight culture to 10mL LB in a sterile shake flask. This was incubated at 37˚C whilst shaking for a further 2 hours to give the cells fresh nutrients and allow them to multiply. This culture was then divided into 3 tubes (3mL in each) and subtilin added in the following concentrations:
| + | |
| - | | + | |
| - | '''- 0% induction''' (3mL dilute iGEMgfp)
| + | |
| - | | + | |
| - | '''- 1% induction''' (3mL dilute iGEMgfp + 30μl subtilin)
| + | |
| | | | |
| - | '''- 10% induction''' (30mL dilute iGEMgfp + 300μl subtilin) | + | * BBa_I746107 contains a partial ''agr'' operon, which includes ''agrC'' and ''A'' but has ''agrB'' and ''D'' deleted. This allows coding for the receptor (''C''/''A'') but not for production of the quorum peptides themselves (''B''/''D''). The inducible promoter drives expression of gfp. |
| | | | |
| - | These were incubated whilst shaking for 1 hour at 37°C. Flow cytometry was then performed on the samples.
| |
| | | | |
| - | * Overnight cultures were made from glycerol stocks pGFPrrnB-ncl08 colony 7 and pJWV021-ncl08 colony 13 (see protocol). The plasmid from these cultures are to be checked for the 2.2kb fragment before being sent for sequencing.
| |
| | | | |
| - | ==Friday 12th September== | + | <html><a name="subcloning"></html> |
| | + | == Subcloning == |
| | | | |
| - | * The ON cultures of iGEMgfp and Bs168 were diluted by adding 250μl ON culture to 10mL LB and incubating at 37°C whilst shaking for 2 hours. The two diluted cultures were then each split into 5 x 2mL aliquots and subtilin added in the following concentrations:
| + | The pUC57 was restriction-digested and the fragments displayed on a gel to ensure that the correct products have been produced. |
| - | - 0% induction (2mL dilute culture)
| + | |
| | | | |
| - | '''- 0.1% induction''' (2mL dilute culture + 2μl subtilin) | + | [[Image:02091.jpg|500px]] |
| | + | Figure 2: Gel from [[Newcastle_University_Wetlab/2_September_2008|2 Sept]] showing our 3 unrestricted plasmids, along with the 4 restrictions |
| | + | * '''Lane 1:''' 1kb ladder |
| | + | * '''Lane 2:''' Unrestricted pGFP-rrnB sample 1 (our bacillus integration vector that carries gfp between 2 amyE flanking regions) |
| | + | * '''Lane 3:''' Unrestricted pGFP-rrnB sample 2 |
| | + | * '''Lane 4:''' Unrestricted pJWV021 sample 1 (our bacillus integration vector that carries mCherry between 2 sacA flanking regions) |
| | | | |
| - | '''- 0.5% induction''' (2Ml dilute culture + 10μl subtilin) | + | * '''Lane 5:''' Unrestricted pJWV021 sample 2 |
| | + | * '''Lane 6:''' Unrestricted pUC57-ncl08 sample 1 (our synthetic biobrick construct; GenScript) |
| | + | * '''Lane 7:''' Unrestricted pUC57-ncl08 sample 2 |
| | + | * '''Lane 8:''' 1kb ladder |
| | + | * '''Lane 9:''' pGFP-rrnB restricted with EcoR1 and Nhe1 |
| | + | * '''Lane 10:''' pJWV021 restricted with BglII and Nhe1 |
| | + | * '''Lane 11:''' pUC57-ncl08 restricted with EcoR1 and Nhe1 |
| | + | * '''Lane 12:''' pUC57-ncl08 restricted with BglII and Nhe1 |
| | | | |
| - | '''- 1% induction''' (2mL dilute culture + 20μl subtilin)
| + | Not shown here is the agr biobrick digestion. |
| | | | |
| - | '''-10% induction''' (2mL dilute culture + 200μl subtilin)
| + | <html><a name="vector-validation"></html> |
| | + | == Validation of the integration vector == |
| | | | |
| - | The five Bs168 tubes acted as controls.
| |
| - | These were incubated at 37°C whilst shaking for 1 hour to allow the subtilin to induce ''gfp'' expression.
| |
| | | | |
| - | * Plasmid was isolated from the 12 pGFPrrnB-BB107 ON cultures 9see protocol). Unfortunately plasmid from colony 4 (25μl plate) and colony 4 (250μl plate) were lost due to human error.
| + | Digested pGFP-rrnB/pJWV021 were mixed with digested pUC57-ncl08 or agr biobrick, ligated and transformed to E.coli and the bacteria [[Newcastle_University_Wetlab/4_September_2008|overnight]] were grown on selective media (the pGFP ligation on plates with spectinomycin (50microG/ml) and the pJWV021 ligation on kanamycin (10 microG/ml)). Transformants were grown up in LB with antibiotics, plasmid was isolated and digested to check for successful subcloning (in the GFP and mCherry vectors, GFP results shown below). |
| | | | |
| - | * pGFPrrnB-ncl08 colony 7 and pJWV021 colony 13 isolated from overnight cultures (see protocol). | + | [[Image:Gel_2.jpg|500px]] |
| | + | Figure 4: Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies |
| | + | * '''Lane 1:''' 1kb ladder |
| | + | * '''Lane 2:''' pGFPrrnB-ncl08 colony 1 (white colony, 25μl plate) |
| | + | * '''Lane 3:''' pGFPrrnB-ncl08 colony 2 (white colony, 25μl plate) |
| | + | * '''Lane 4:''' pGFPrrnB-ncl08 colony 3 (white colony, 25μl plate) |
| | + | * '''Lane 5:''' pGFPrrnB-ncl08 colony 4 (white colony, 25μl plate) |
| | + | * '''Lane 6:''' pGFPrrnB-ncl08 colony 5 (white colony, 25μl plate) |
| | + | * '''Lane 7:''' pGFPrrnB-ncl08 colony 6 (white colony, 25μl plate) |
| | + | * '''Lane 8:''' pGFPrrnB-ncl08 colony 7 (white colony, 25μl plate) |
| | + | * '''Lane 9:''' pGFPrrnB-ncl08 colony 8 (white colony, 25μl plate) |
| | + | * '''Lane 10:''' pGFPrrnB-ncl08 colony 9 (white colony, 25μl plate) |
| | + | * '''Lane 11:''' pGFPrrnB-ncl08 colony 10 (white colony, 250μl plate) |
| | + | * '''Lane 12:''' pGFPrrnB-ncl08 colony 11 (white colony, 250μl plate) |
| | + | * '''Lane 13:''' pGFPrrnB-ncl08 colony 12 (green colony, 25μl plate) |
| | | | |
| - | * All isolated plasmid was restricted using 7.5μl plasmid and 0.5μl each enzyme in 15μl total volume:
| + | This demonstrated that we had succesfully subcloned our ncl08 biobrick from pUC57-ncl08 to pGFP. Not shown here are the restriction checks for pJWV021-ncl08 and pGFP-agr (but we have correct clones for them as well!). |
| - | - pGFPrrnB-BB107 restrited with EcoRI and SpeI. | + | |
| | | | |
| - | - pGFPrrnB-ncl08 restricted with EcoRI and NheI
| |
| | | | |
| - | - pJWV021-ncl08 restricted with NheI and BglII | + | <html><a name="bsubtilis-integration"></html> |
| | + | ==Transformation and integration into the ''B. subtilis'' chromosome== |
| | | | |
| - | These restrictions were run on gels.
| + | [[Image:CloningStratStep2.jpg|500px|thumb|left]] |
| | | | |
| - | '''Lane 1:''' 1kb ladder
| |
| | | | |
| - | '''Lane 2:''' pGFPrrnB-BB107 colony 1
| + | <html><a name="chromosome-validation"></html> |
| | + | ==Validation of chromosomal integration== |
| | | | |
| - | '''Lane 3:''' pGFPrrnB-BB107 colony 2 | + | Next, we transformed the correct plasmids to (self made) competent ''Bacillus'', as described in the protocol section. For plasmid pGFP-ncl08 and pGFP-agr, bacillus was plated on chloramphenicl (5 microG/ml) and for plasmid pJWV021-ncl08, bacillus were plated on kanamycin (5 microG/ml). Next day, Bacillus transformants were subseqently streaked to single colonies on selective agar plates. To check for correct integration of the pGFP-ncl08 and pGFP-agr plasmids at the amyE locus, single colonies were streaked on nutrient agar plates containing 1% starch. If a double crossover event occured, cells are not able to produce AmyE and will not degrade starch, thus no 'halo' around the colony. 2 individual colonies without an halo were grown and stocked and used for later analyses. To check correct integration of the pJWV021-ncl08 plasmid in the Bacillus chromosome, single colonies were streaked on minimal medium plates containing either glucose or sucrose as the sole carbon source. If the plasmid integrated correctly at the sacA locus via a double crossover, cells cannot utilise sucrose anymore and will not grow on the minimal plate containing sucrose, but will grow on the glucose plate. Again, 2 correct transformants were selected that were unable to proliferate on sucrose plates and stocked in the -80C freezer for future work. |
| | | | |
| - | '''Lane 4:''' pGFPrrnB-BB107 colony 3
| |
| | | | |
| - | '''Lane 5:''' pGFPrrnB-BB107 colony 5
| |
| | | | |
| - | '''Lane 6:''' pGFPrrnB-BB107 colony 6
| + | <html><a name="characterisation"></html> |
| | + | ==Characterisation of the integrated parts and their response to subtilin== |
| | | | |
| - | '''Lane 7:''' pGFPrrnB-BB107 colony 7
| + | <html><a name="results"></html> |
| | + | == Results == |
| | | | |
| - | '''Lane 8:''' pGFPrrnB-BB107 colony 8 | + | To analyse the results of the wet lab transformations of the inserts into ''B. subtilis'', we used two methods: microscopy and flow cytometry. |
| | | | |
| - | '''Lane 9:''' pGFPrrnB-BB107 colony 9
| + | ===Microscopy=== |
| | | | |
| - | '''Lane 10:''' pGFPrrnB-BB107 colony 10 | + | Microscopy work from 08.09.08 showed a difference in the level of flourescence of the iGEMgfp fluorescent cells (higher in 10% subtilin-induced cells compared to 0% subtilin-induced cells). However, there was little difference in the ''number'' of cells that fluoresced between the two cultures. |
| | | | |
| - | '''Lane 11:''' pGFPrrnB-BB107 colony 11 | + | There was no difference in the number of fluorecent cells ''or'' the level of fluorescence between the 10% subtilin-induced and the 0% subtilin-induced iGEMcherry cells. |
| | | | |
| | | | |
| - | '''Lane 1:''' 1kb ladder
| + | ===Flow cytometry=== |
| | | | |
| - | '''Lane 2:''' pGFPrrnB-ncl08 colony 7 | + | Flow cytometry measures cell fluorescence and light scattering, cell-by-cell, allowing us to quantify our results and present them in graphical form. A sample of our cells engineered ''B. subtilis'' cells were injected into the machine, which hydro-dynamically focusses the fluid. Lasers of a particular wavelength are directed onto the stream of fluid, and each particle which passes through the light beam will cause the laser to scatter in a different way. Cells which absorb and then re-emit the light, result in fluorescence (a higher energy state). |
| | | | |
| - | '''Lane 3:''' pJWV021-ncl08 colony 13
| + | The detectors in the machine measure the scattering of light and any fluoresence which occurs, and the data is displayed as histograms and dot plots. |
| | | | |
| - | '''Lane 4:''' pGFPrrnB-ncl08 colony 7 control (no enzyme) | + | '''Induction by 0% v/v subtilin''' (i.e in the absence of subtilin): mean fluoresence = 7.70 |
| | | | |
| - | '''Lane 5:''' pJWV021-ncl08 colony 13 control (no enzyme) | + | '''Induction by 1% v/v subtilin:''' mean fluoresence = 14.77 |
| | | | |
| - | ==Monday 15th September== | + | '''Induction by 10% v/v subtilin:''' the mean fluoresence = 21.95 |
| | | | |
| - | * ON cultures were made from [[glycerol stocks]] of pGFPrrnB-ncl08 colonies 7 + 11 and pGFPrrnB-BB107 colonies 3 + 5.
| + | These results show that the higher the concentration of subtilin, the more GFP is expressed. |
| | | | |
| - | * [[ON culture]] made from ''B. subtilis'' ON culture:
| + | At 10% induction, the subtilin began to cause some of the cells to lyse - this can be seen by the two distinct populations of cells - one being the cells which have lysed, and the other being the remainder of intact cells. Since subtilin is an antibiotic, this was a good indication that the subtilin we had collected that day was of good quality. |
| - | </div><!--maincontent -->
| + | |
| | | | |
| - | ==Tuesday 16th September==
| + | Overall, the microscopy and flow cytometry suggest that our biobrick is functional, but requires high levels of subtilin in order to be activated. The days that we performed microscopy for instance, we did not observe killing of the cells after 10% addition of subtilin, indicating that our subtilin for that day was at a low concentration. Subsequently, we did not observe a strong GFP induction. However, when we did observe killing (indicative of high subtilin concentrations), we did observe induction of GFP (see flow cytometry). So in principle the system works, and we have engineered the lab strain of ''B. subtilis'' 168 to respond to the external addition of subtilin, but more work should be done to further optimise the system. For instance, it was shown that the spaS promoter fused to gfp gives high yield of protein production when present on a multicopy plasmid, after the addition of subtilin (Bongers et al, AEM). Also it may be possible to introduce a positive feedback effect by introducing a ''spaS'' box and associated promoter upstream of ''spaRK'' in addition to the normal promoter. |
| - | [[Image:16091.jpg|thumb|right|300px|Gel showing pGFPrrnB-ncl08 and pGFPrrnB-BBa_...107 restrictions from different colonies.]]
| + | |
| | | | |
| - | * Plasmid [[isolated]] from pGFPrrnB-ncl08 colonies 7 + 11 and pGFPrrnB-BB107 colonies 3 + 5 ON cultures.
| + | <gallery> |
| | + | Image:Montage.jpg|Microscopy results of gfp upon 1% induction by subtilin |
| | + | Image:Montage_pc.jpg|Microscopy results of mCherry upon 1% induction by subtilin |
| | + | Image:Flow0%.jpg|Flow cytometry results for 0% induction by subtilin. |
| | + | Image:Flow1%.jpg|Flow cytometry results for 1% induction by subtilin. |
| | + | Image:Flow10%.jpg|Flow cytometry results for 10% induction by subtilin. |
| | + | </gallery> |
| | | | |
| - | * A Sample of each of the 4 isolated plasmids were [[restricted]] using 7.5μl plasmid and 0.5μl each enzyme in 15μl total volume:
| |
| | | | |
| - | - pGFPrrnB-ncl08 restricted with EcoRI + NheI
| |
| | | | |
| - | - pGFPrrnB-BB107 restricted with EcoRI + SpeI
| + | <html><a name="protocols"></html> |
| | + | ===Protocols=== |
| | | | |
| - | Gel:
| + | Listed below are the lab protocols that were developed or used for the Newcastle University iGEM 2008 wet lab component. Please click on any item for detailed instructions. |
| | | | |
| - | '''Expected band sizes:'''
| + | * [[Agarose Gel Electrophoresis]] |
| - | * '''Lane 2:''' ~ 8.4kb and 2.2kb
| + | * [[Making Agar Plates]] |
| - | * '''Lane 3:''' ~ 8.4kb and 2.2kb
| + | * [[Isolating Plasmid from Cells (Miniprep)]] |
| - | * '''Lane 4:''' ~ 8.4kb and 3.2kb
| + | * [[Restricting Plasmids (Double Restriction)]] |
| - | * '''lane 5:''' ~ 8.4kb and 3.2kb
| + | * [[Purifying DNA from an enzymatic reaction]] |
| | + | * [[Cutting a Specific Band from Agarose Gel]] |
| | + | * [[Purifying DNA from Gel Slices]] |
| | + | * [[Ligating DNA]] |
| | + | * [[Transforming into DH5α or TOP10 E. coli|Transforming into DH5α or TOP10 ''E. coli'']] |
| | + | * [[Making Frozen Glycerol Cell Stocks From Overnight Cultures]] |
| | + | * [[Making Overnight Cultures from Frozen Glycerol Cell Stocks]] |
| | + | * [[Making Overnight Cultures from Agar Colonies]] |
| | + | * [[Transforming into Bacillus subtillis|Transforming into ''Bacillus subtilis'']] |
| | + | * [[Inducing Bacillus subtilis with Subtilin|Inducing ''Bacillus subtilis'' with Subtilin]] |
| | + | * [[Preparing Bacillus subtilis for Microscopy|Preparing ''Bacillus subtilis'' for Microscopy]] |
| | | | |
| - | '''Lane 1:''' 1kb ladder
| |
| | | | |
| - | '''Lane 2:''' pGFPrrnB-ncl08 colony 7 restricted with EcoR1 and Nhe1
| |
| | | | |
| - | '''Lane 3:''' pGFPrrnB-ncl08 colony 11 restricted with EcoR1 and Nhe1
| + | <html><a name="journal"></html> |
| | + | == Wet Lab Journal == |
| | | | |
| - | '''Lane 4:''' pGFPrrnB-BBa_...107 colony 3 restricted with EcoR1 and Spe1
| + | This contains the day-to-day progress of our wet lab team. Click on a day below to find more details of the work above. |
| | | | |
| - | '''lane 5:''' pGFPrrnB-BBa_...107 colony 5 restricted with EcoR1 and Spe1
| + | {|cellpadding="10" |
| | + | |{{#calendar: title=Newcastle University Wetlab |year=2008 | month=08 }} |
| | + | |{{#calendar: title=Newcastle University Wetlab |year=2008 | month=09 }} |
| | + | |} |
| | | | |
| - | As you can see from our gel, all 4 restrictions yielded the expected sized bands, indicating that out ligations had been successful. The remainder of the unrestricted plasmid from pGFPrrnB-ncl08 colony 7 was stored in the -20 freezer ready to be sent off for sequencing. The two unrestricted pGFPrrnb-BBa_...107 plasmid samples were then transformed into B.subtilis 168.
| + | </div><!--main col --> |
Introduction
The overall design was too complicated for the time and resources available so we devised a proof of concept Brick which represented an input node from our full design. This Brick demonstrates that it is possible to transfer two-component peptide sensing from one strain into another and have it function as it did in the first strain. We took the subtilin two component system from Bacillus subtilis ATCC6633 and integrated it into the chromosome of B. subtilis 168, the typical laboratory B. subtilis strain at the amyE locus.
The wet lab work involved:
- Cloning the synthesized Brick from pUC57 in E. coli into our Bacillus integration vectors (with GFP and mCherry respectively downstream of the Brick)
- Validation of the integration vector
- Transformation and integration into the chromosome of B. subtilis 168 at the amyE locus
- Validation of chromosomal integration
- Characterisation of the integrated parts and their response to subtilin.
Our subtilin sensor, part [http://partsregistry.org/Part:BBa_K104001 BBa_k104001]

Figure 1: Newcastle's iGEM team construct.
Figure 1 shows the construct which contains:
- spaRK promotor
- spaR (subtilin peptide antibiotic Regulation) - the 220 amino acid product of this gene usually regulates the downstream production of subtilin antibiotic. It has an N-terminal domain that can be phosphorylated and a C-terminal domian that has DNA binding properties [http://aem.asm.org/cgi/reprint/59/1/296.pdf]
- spaK (subtilin peptide antibiotic Kinase) - this gene codes for a 325 amino acid histadine kinase peptide that phosphorylates the N-terminus of spaR [http://aem.asm.org/cgi/reprint/59/1/296.pdf]. This activates the DNA binding ability of the C-terminus of spaR, which in turn initiates transcription of the downstream gene. In the case of our construct, this gene is gfp.
- rrnO transcriptional terminator
- spaS promotor - a strong promotor inducible by upstream activation of spaRK. It can be placed in front any gene to regulate its activity.
Aim 1: clone the spaRK system from pUC57 into pJWV021 and transform into DH5 alpha competent E. coli
Aim 2: clone the spaRK system from pUC57 into pGFP-rrnB and transform into DH5 alpha competent E. coli
- The 2.2kb fragment (ncl08) contains the spaRK biobrick system. When properly cloned, our biobrick will replace the rrnB promoter in front of gfp present in plasmid pGFP-rrnB. Thus, gfp will now be under control of the subtilin inducible spaS promoter. This means that when SpaR is activated by SpaK (by sensing subtilin) its positive regulatory effect on PspaS will activate gfp.
Aim 3: clone the agr (Cambridge biobrick for sensing S. aureus) system from the biobrick plasmid using EcoRI and SpeI sites into the corresponding sites of pGFP-rrnB. This will replace the PrrnB-gfp on the bacillus vector with the agr biobrick. The ligation was transformed into DH5 alpha competent E. coli
- BBa_I746107 contains a partial agr operon, which includes agrC and A but has agrB and D deleted. This allows coding for the receptor (C/A) but not for production of the quorum peptides themselves (B/D). The inducible promoter drives expression of gfp.
Subcloning
The pUC57 was restriction-digested and the fragments displayed on a gel to ensure that the correct products have been produced.
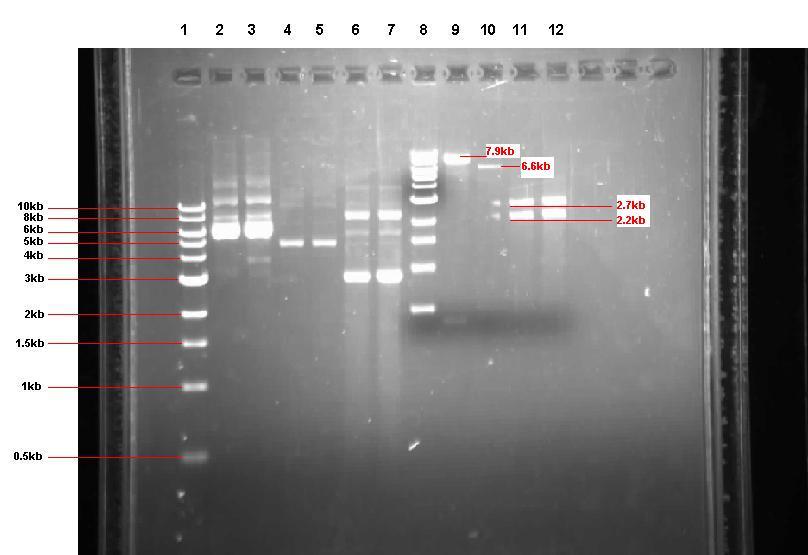 Figure 2: Gel from 2 Sept showing our 3 unrestricted plasmids, along with the 4 restrictions
Figure 2: Gel from 2 Sept showing our 3 unrestricted plasmids, along with the 4 restrictions
- Lane 1: 1kb ladder
- Lane 2: Unrestricted pGFP-rrnB sample 1 (our bacillus integration vector that carries gfp between 2 amyE flanking regions)
- Lane 3: Unrestricted pGFP-rrnB sample 2
- Lane 4: Unrestricted pJWV021 sample 1 (our bacillus integration vector that carries mCherry between 2 sacA flanking regions)
- Lane 5: Unrestricted pJWV021 sample 2
- Lane 6: Unrestricted pUC57-ncl08 sample 1 (our synthetic biobrick construct; GenScript)
- Lane 7: Unrestricted pUC57-ncl08 sample 2
- Lane 8: 1kb ladder
- Lane 9: pGFP-rrnB restricted with EcoR1 and Nhe1
- Lane 10: pJWV021 restricted with BglII and Nhe1
- Lane 11: pUC57-ncl08 restricted with EcoR1 and Nhe1
- Lane 12: pUC57-ncl08 restricted with BglII and Nhe1
Not shown here is the agr biobrick digestion.
Validation of the integration vector
Digested pGFP-rrnB/pJWV021 were mixed with digested pUC57-ncl08 or agr biobrick, ligated and transformed to E.coli and the bacteria overnight were grown on selective media (the pGFP ligation on plates with spectinomycin (50microG/ml) and the pJWV021 ligation on kanamycin (10 microG/ml)). Transformants were grown up in LB with antibiotics, plasmid was isolated and digested to check for successful subcloning (in the GFP and mCherry vectors, GFP results shown below).
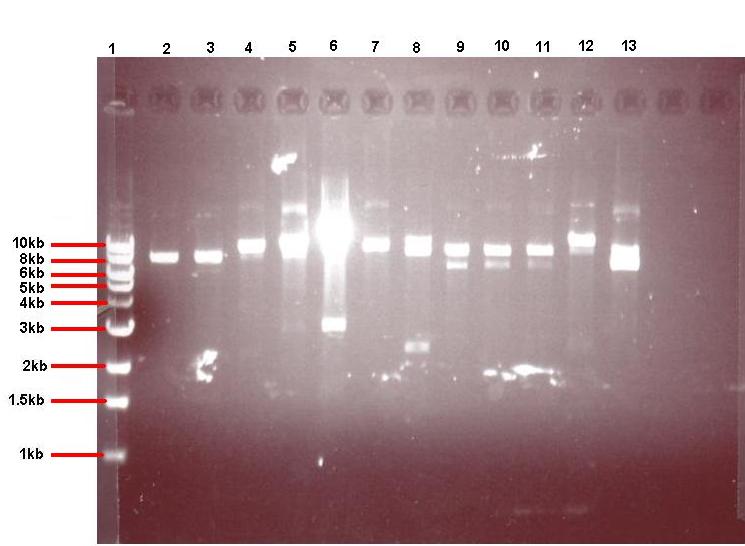 Figure 4: Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies
Figure 4: Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies
- Lane 1: 1kb ladder
- Lane 2: pGFPrrnB-ncl08 colony 1 (white colony, 25μl plate)
- Lane 3: pGFPrrnB-ncl08 colony 2 (white colony, 25μl plate)
- Lane 4: pGFPrrnB-ncl08 colony 3 (white colony, 25μl plate)
- Lane 5: pGFPrrnB-ncl08 colony 4 (white colony, 25μl plate)
- Lane 6: pGFPrrnB-ncl08 colony 5 (white colony, 25μl plate)
- Lane 7: pGFPrrnB-ncl08 colony 6 (white colony, 25μl plate)
- Lane 8: pGFPrrnB-ncl08 colony 7 (white colony, 25μl plate)
- Lane 9: pGFPrrnB-ncl08 colony 8 (white colony, 25μl plate)
- Lane 10: pGFPrrnB-ncl08 colony 9 (white colony, 25μl plate)
- Lane 11: pGFPrrnB-ncl08 colony 10 (white colony, 250μl plate)
- Lane 12: pGFPrrnB-ncl08 colony 11 (white colony, 250μl plate)
- Lane 13: pGFPrrnB-ncl08 colony 12 (green colony, 25μl plate)
This demonstrated that we had succesfully subcloned our ncl08 biobrick from pUC57-ncl08 to pGFP. Not shown here are the restriction checks for pJWV021-ncl08 and pGFP-agr (but we have correct clones for them as well!).
Transformation and integration into the B. subtilis chromosome
Validation of chromosomal integration
Next, we transformed the correct plasmids to (self made) competent Bacillus, as described in the protocol section. For plasmid pGFP-ncl08 and pGFP-agr, bacillus was plated on chloramphenicl (5 microG/ml) and for plasmid pJWV021-ncl08, bacillus were plated on kanamycin (5 microG/ml). Next day, Bacillus transformants were subseqently streaked to single colonies on selective agar plates. To check for correct integration of the pGFP-ncl08 and pGFP-agr plasmids at the amyE locus, single colonies were streaked on nutrient agar plates containing 1% starch. If a double crossover event occured, cells are not able to produce AmyE and will not degrade starch, thus no 'halo' around the colony. 2 individual colonies without an halo were grown and stocked and used for later analyses. To check correct integration of the pJWV021-ncl08 plasmid in the Bacillus chromosome, single colonies were streaked on minimal medium plates containing either glucose or sucrose as the sole carbon source. If the plasmid integrated correctly at the sacA locus via a double crossover, cells cannot utilise sucrose anymore and will not grow on the minimal plate containing sucrose, but will grow on the glucose plate. Again, 2 correct transformants were selected that were unable to proliferate on sucrose plates and stocked in the -80C freezer for future work.
Characterisation of the integrated parts and their response to subtilin
Results
To analyse the results of the wet lab transformations of the inserts into B. subtilis, we used two methods: microscopy and flow cytometry.
Microscopy
Microscopy work from 08.09.08 showed a difference in the level of flourescence of the iGEMgfp fluorescent cells (higher in 10% subtilin-induced cells compared to 0% subtilin-induced cells). However, there was little difference in the number of cells that fluoresced between the two cultures.
There was no difference in the number of fluorecent cells or the level of fluorescence between the 10% subtilin-induced and the 0% subtilin-induced iGEMcherry cells.
Flow cytometry
Flow cytometry measures cell fluorescence and light scattering, cell-by-cell, allowing us to quantify our results and present them in graphical form. A sample of our cells engineered B. subtilis cells were injected into the machine, which hydro-dynamically focusses the fluid. Lasers of a particular wavelength are directed onto the stream of fluid, and each particle which passes through the light beam will cause the laser to scatter in a different way. Cells which absorb and then re-emit the light, result in fluorescence (a higher energy state).
The detectors in the machine measure the scattering of light and any fluoresence which occurs, and the data is displayed as histograms and dot plots.
Induction by 0% v/v subtilin (i.e in the absence of subtilin): mean fluoresence = 7.70
Induction by 1% v/v subtilin: mean fluoresence = 14.77
Induction by 10% v/v subtilin: the mean fluoresence = 21.95
These results show that the higher the concentration of subtilin, the more GFP is expressed.
At 10% induction, the subtilin began to cause some of the cells to lyse - this can be seen by the two distinct populations of cells - one being the cells which have lysed, and the other being the remainder of intact cells. Since subtilin is an antibiotic, this was a good indication that the subtilin we had collected that day was of good quality.
Overall, the microscopy and flow cytometry suggest that our biobrick is functional, but requires high levels of subtilin in order to be activated. The days that we performed microscopy for instance, we did not observe killing of the cells after 10% addition of subtilin, indicating that our subtilin for that day was at a low concentration. Subsequently, we did not observe a strong GFP induction. However, when we did observe killing (indicative of high subtilin concentrations), we did observe induction of GFP (see flow cytometry). So in principle the system works, and we have engineered the lab strain of B. subtilis 168 to respond to the external addition of subtilin, but more work should be done to further optimise the system. For instance, it was shown that the spaS promoter fused to gfp gives high yield of protein production when present on a multicopy plasmid, after the addition of subtilin (Bongers et al, AEM). Also it may be possible to introduce a positive feedback effect by introducing a spaS box and associated promoter upstream of spaRK in addition to the normal promoter.
Microscopy results of gfp upon 1% induction by subtilin
|
Microscopy results of mCherry upon 1% induction by subtilin
|
Flow cytometry results for 0% induction by subtilin.
|
Flow cytometry results for 1% induction by subtilin.
|
Flow cytometry results for 10% induction by subtilin.
|
Protocols
Listed below are the lab protocols that were developed or used for the Newcastle University iGEM 2008 wet lab component. Please click on any item for detailed instructions.
Wet Lab Journal
This contains the day-to-day progress of our wet lab team. Click on a day below to find more details of the work above.
| August
|
| M | T | W | T | F | S | S
|
|
|
|
|
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/1_August_2008&action=edit 1]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/2_August_2008&action=edit 2]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/3_August_2008&action=edit 3]
|
| [http://2008.igem.org/Newcastle_University_Wetlab/4_August_2008 4]
| [http://2008.igem.org/Newcastle_University_Wetlab/5_August_2008 5]
| [http://2008.igem.org/Newcastle_University_Wetlab/6_August_2008 6]
| [http://2008.igem.org/Newcastle_University_Wetlab/7_August_2008 7]
| [http://2008.igem.org/Newcastle_University_Wetlab/8_August_2008 8]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/9_August_2008&action=edit 9]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/10_August_2008&action=edit 10]
|
| [http://2008.igem.org/Newcastle_University_Wetlab/11_August_2008 11]
| [http://2008.igem.org/Newcastle_University_Wetlab/12_August_2008 12]
| [http://2008.igem.org/Newcastle_University_Wetlab/13_August_2008 13]
| [http://2008.igem.org/Newcastle_University_Wetlab/14_August_2008 14]
| [http://2008.igem.org/Newcastle_University_Wetlab/15_August_2008 15]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/16_August_2008&action=edit 16]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/17_August_2008&action=edit 17]
|
| [http://2008.igem.org/Newcastle_University_Wetlab/18_August_2008 18]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/19_August_2008&action=edit 19]
| [http://2008.igem.org/Newcastle_University_Wetlab/20_August_2008 20]
| [http://2008.igem.org/Newcastle_University_Wetlab/21_August_2008 21]
| [http://2008.igem.org/Newcastle_University_Wetlab/22_August_2008 22]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/23_August_2008&action=edit 23]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/24_August_2008&action=edit 24]
|
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/25_August_2008&action=edit 25]
| [http://2008.igem.org/Newcastle_University_Wetlab/26_August_2008 26]
| [http://2008.igem.org/Newcastle_University_Wetlab/27_August_2008 27]
| [http://2008.igem.org/Newcastle_University_Wetlab/28_August_2008 28]
| [http://2008.igem.org/Newcastle_University_Wetlab/29_August_2008 29]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/30_August_2008&action=edit 30]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/31_August_2008&action=edit 31]
|
|
| September
|
| M | T | W | T | F | S | S
|
| [http://2008.igem.org/Newcastle_University_Wetlab/1_September_2008 1]
| [http://2008.igem.org/Newcastle_University_Wetlab/2_September_2008 2]
| [http://2008.igem.org/Newcastle_University_Wetlab/3_September_2008 3]
| [http://2008.igem.org/Newcastle_University_Wetlab/4_September_2008 4]
| [http://2008.igem.org/Newcastle_University_Wetlab/5_September_2008 5]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/6_September_2008&action=edit 6]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/7_September_2008&action=edit 7]
|
| [http://2008.igem.org/Newcastle_University_Wetlab/8_September_2008 8]
| [http://2008.igem.org/Newcastle_University_Wetlab/9_September_2008 9]
| [http://2008.igem.org/Newcastle_University_Wetlab/10_September_2008 10]
| [http://2008.igem.org/Newcastle_University_Wetlab/11_September_2008 11]
| [http://2008.igem.org/Newcastle_University_Wetlab/12_September_2008 12]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/13_September_2008&action=edit 13]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/14_September_2008&action=edit 14]
|
| [http://2008.igem.org/Newcastle_University_Wetlab/15_September_2008 15]
| [http://2008.igem.org/Newcastle_University_Wetlab/16_September_2008 16]
| [http://2008.igem.org/Newcastle_University_Wetlab/17_September_2008 17]
| [http://2008.igem.org/Newcastle_University_Wetlab/18_September_2008 18]
| [http://2008.igem.org/Newcastle_University_Wetlab/19_September_2008 19]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/20_September_2008&action=edit 20]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/21_September_2008&action=edit 21]
|
| [http://2008.igem.org/Newcastle_University_Wetlab/22_September_2008 22]
| [http://2008.igem.org/Newcastle_University_Wetlab/23_September_2008 23]
| [http://2008.igem.org/Newcastle_University_Wetlab/24_September_2008 24]
| [http://2008.igem.org/Newcastle_University_Wetlab/25_September_2008 25]
| [http://2008.igem.org/Newcastle_University_Wetlab/26_September_2008 26]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/27_September_2008&action=edit 27]
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/28_September_2008&action=edit 28]
|
| [http://2008.igem.org/wiki/index.php?title=Newcastle_University_Wetlab/29_September_2008&action=edit 29]
| [http://2008.igem.org/Newcastle_University_Wetlab/30_September_2008 30]
|
|
 "
"




 Figure 2: Gel from 2 Sept showing our 3 unrestricted plasmids, along with the 4 restrictions
Figure 2: Gel from 2 Sept showing our 3 unrestricted plasmids, along with the 4 restrictions
 Figure 4: Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies
Figure 4: Gel showing restricted pGFPrrnB-ncl08 taken from 12 different colonies





