|
_DNA-Origami
Introduction
Paul Rothemund has discovered that it is possible to shape M13-Phage single-strand-DNA simply adding oligonucleotides that will work as „brackets“ when complementing the long single-strand.
In this way, one can generate for example DNA-squares of a certain size with „nods“ at certain distances.
One member of our team, Daniel Hautzinger, has recently finished his diploma-thesis on Origami-DNA and the possibilities of generating patterns on these square surfaces by modifying the oligo-nucleotides that build up the nod-points.
As the antigens NIP and fluoresceine can as well be fused to these oligos, we had found the seemingly perfect tool to present strictly defined two-dimensional antigen-patterns to cells carrying our synthetic receptor system.
Methods
Phage DNA
Cell culture
50 ml DYT-Medium, 50 µl tetracycline (TET; 25 mg/ml) and ER2738-cells
were shaken over night at 37°C. The overnight culture was diluted with
DYT to OD600=0.1 and shaken at 37°C until the culture reached an OD600
around 0.4. Each 50 ml of cell culture were inoculated with 5 µl
M13mp18 phage and shaken for 4 h at 37°C.
Isolation of M13mp18 phage from cell culture
PEG/NaCl was used to precipitate the phages.
First precipitation
Each 50 ml of cell culture was centrifuged at 5000 g for 20 min. While
the cells were centrifuged, 1/7 volume PEG/NaCl (about 7ml) was added and
transferred in a falcon tube. The phages stay in the
supernatant therefore the supernatant was carefully decanted to the
PEG/NaCl and mixed gently by inverting the tube. The mixture (solution
1) was left overnight at 4°C.
Solution 1 was centrifuged at 5000 g for 20 min. Because the phage stay
in the pellet, the supernatant was removed and the pellet was
resuspended in 2 ml TBS-Buffer (solution 2). Solution 2 was put in a
1.5 ml Eppendorf tube and centrifuged (13200 rpm, 10 min). After the
centrifugation the phages stay in the supernatant.
Second precipitation
170 µl PEG/NaCl(~ 1/6 volume of supernatant) were put in a Eppendorf
tube. Supernatant was carefully decanted to the PEG/NaCl and mixed
gently by inverting the tube. The mixture (solution 3) was left for 1 h
on ice. Solution 3 was centrifuged at 13200 rpm for 10 min.
Define phage titers
The absorption of solution 3 was measured on a Jasco V-550 UV/VIS
spectrometer at 269 nm.
Phage titer was calculated as follows:
Phage DNA =
((A269-A360) * 6 * 10^16 * dilution factor) / (number of bases in the
phage genom = 7249 bp)
Isolation of the phage DNA
The phage DNA was isolated with QIAprep Spin M13-Kit (50) from QIAGEN
(Cat.No: 27704).
DNA-concentration was quantified by Nano-drop photometer.
Origami
Produce
the Origami
To produce the Origami we mixed each the M13mp18 DNA with the oligos,
water and TEA/MgAcetat (end concentration =12.5mM).
Various samples were
produced. For the sample with a ratio of 1:20 (DNA:Oligo), we used 4 nM
DNA and 80 nM of oligos. The Origami were produced in an eppendorf
Mastercycler personal. They were heated up to 95°C for 7 min
and slowly cooled down (0.3°C/s) to 20°C.
Different sample were made:
Sample 1:20, 1:10 and 1:5 without NIP, oligos without NIP were used
Sample 1:20, 1:10 and 1:5 with NIP, all of the 7 oligos with NIP were
used -> origami with 7NIP
Sample 1:5 with NIP and fluorophor, all of the 7 oligos with NIP and
the 2 oligos with the Alexa 488 were used.
Purification of the DNA-Origamis
To purify the DNA-Origamis from the unbound DNA-oligos, we used Montage® PCR Centrifugal Filter Devices (Millipore). The Montage® PCR Centrifugal Filter Devices were labeled and put with the purple side on top in 1.5 ml Eppendorf tubes. To clean the filter of remaining glycerol, 450 µl TAE/MgAcetat (12.5 mM; 1x filtered) was put on top of the filter and centrifuged for 15 min at 1000 g. After removing the filtrate, 400 µl TEA/MgAcetat (12.5 mM;1x filtered) and 45 µl DNA-origami were put on top of the filter and again centrifuged for 15 min at 1000 g. The filtrate was removed again. All unbound DNA-oligos were washed off by putting 400 µl TEA/MgAcetat (12.5 mM; 1x filtered) on top of the filter. The sample was centrifuged for 15 min at 1000 g. To release the DNA-origamis of the filter, 100 µl TAE/MgAcetat (12.5 mM;1x filtered) was put on top of the filter, and the filter was left at room temperature for at least 2 min. The filter shouldn´t run dry. The Montage® PCR Centrifugal Filter Devices were put upside down (the purple side has to be on the bottom) in one of the special Invert Spin tubes form Millipore and centrifuged for 3 min at 1000 g.
The Origami were kept in different buffers. For this, TEA/MgAcetat (12.5 mM; 1x filtered) was replaced by the according buffer.
Atomic force microscopy to prove if the origami stable
To see if the Origami were formed well and stable in the different buffers an atomic force microscope (AFM) was used. The measurement itself was done in air (not in the buffer).
The DNA-Origami were absorbed to freshly cleaved mica. Therefore the mica was cut into 6 mm pieces and affixed to the metal panes we used for the measurement. To get an atomically clean surface, an adhesive tape was used to remove the topmost mica layers. After this the sample could be put on. First 2-10 µl of the sample were put on the mica and then quickly diluted with water (just as much that the mica was covered with fluid). The sample was incubated for about 5 min and then the mica was blown dry with a stream of nitrogen. Then the sample could be measured.
The metal pane was fixed in the metal sample holder by a magnet, so that the sample could not move itself during the measurement.
Results and Discussion
1. Different ratios of Origami
Because we also wanted to measure the calcium influx in the LSRII
fluorescence spectrometer, we had to increase the concentration of the
Origamis at least up to 200 nM. Because the oligos we had to use for
building the Origamis are very expensive, we first tried to reduce the
ratio of Origami to oligos. Therefore we tried to make Origamis with two
(1:10) and four (1:5) times lower concentration of oligos. As positive
control we took the 1:20 ratio at which we knew it should be stable. We
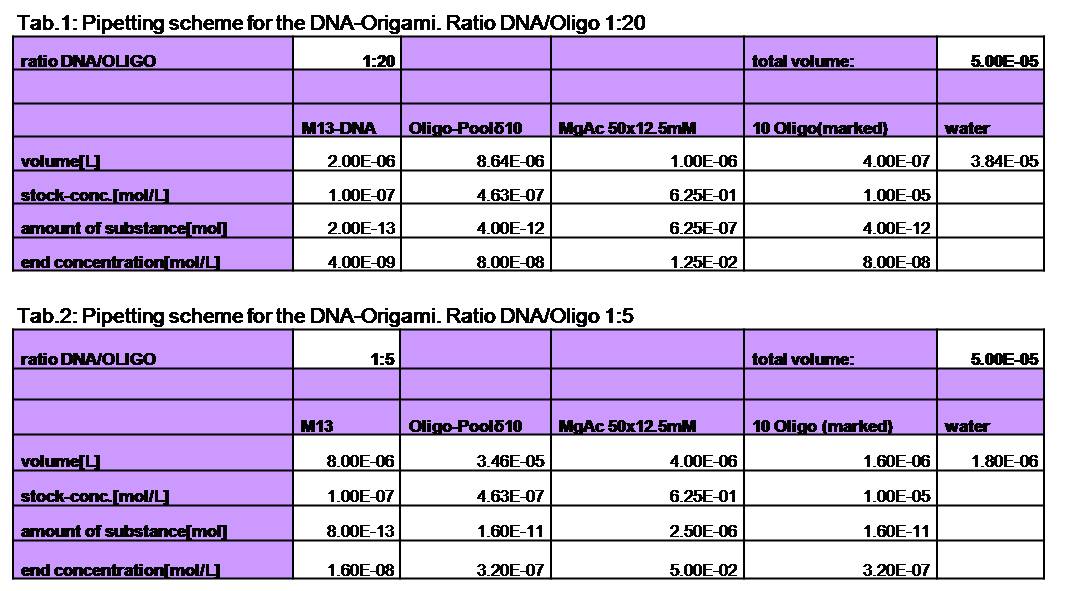
used the AFM to check if the Origami are well formed. The results are
shown in figures 1-3.
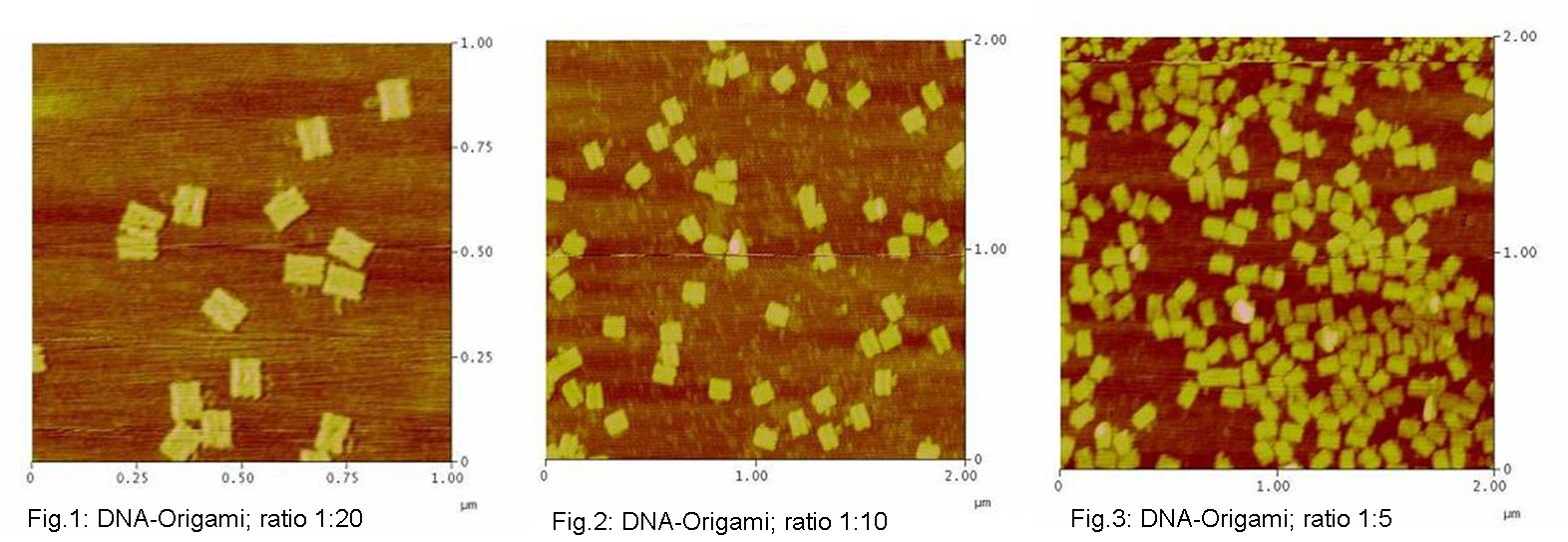
As we see in the Figures 1-3, all Origami are well formed. So we are
able to use a 1:5 ratio to produce our Origami.
2. Origami in Krebs-Ringer-Hepes buffer
The TEA/MgAcetat buffer we used to build and keep the Origami does not
have any salts beside the magnesium. Therefore the T cells are not able
to survive long enough to measure the calcium influx.
Beside that, the EDTA would disturb the calcium measurement. Hence we
had to find a different buffer in which the Origami and the T
cells are stable and which can also be used for the calcium
measurement. In literature we found, that many people use a so-called
Krebs-Ringer-HEPES buffer for calcium measurement in LSRII fluorescence
spectrometer. We used 1:20 and 1:5 ratio of DNA to oligo to
make the Origami. Each of the ratios was buffered in Krebs-ringer-HEPES
buffer. As positive control we used Origami buffered in TAE/MgAc. The
results are shown in figure 4 and 5.
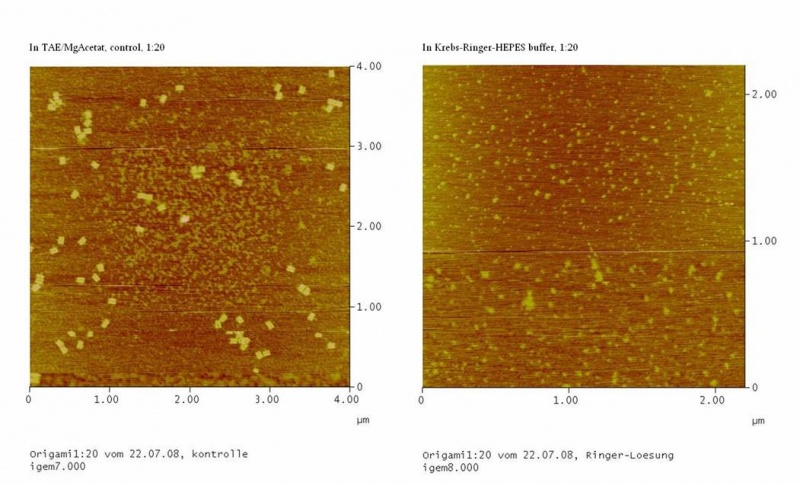
Fig. 4:DNA-Origami(1:20) in TAE/MgAcetat (control) in Krebs-Ringer-Hepes buffer
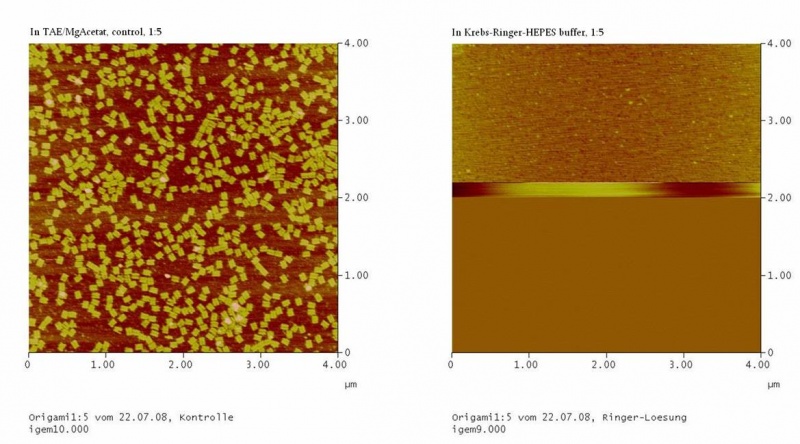 Fig. 5:DNA-Origami(1:5) in TAE/MgAcetat (control) in Krebs-Ringer-Hepes buffer
Fig. 5:DNA-Origami(1:5) in TAE/MgAcetat (control) in Krebs-Ringer-Hepes buffer
Both samples in Krebs-Ringer-HEPES buffer did not show the completely
formed Origami. Some bigger structures in Krebs-Ringer-HEPES buffer in
figure 4 seem to be parts of the Origami. Maybe the Origami did form
right in the MasterCycler, but then fell apart when we buffered them in
the Krebs-Ringer-HEPES buffer. Because we read in literature that some
salts in the Krebs-Ringer-HEPES buffer could disturb the interaction of
the Origami to the glimmer we first incubated the surface of the
glimmer with Krebs-Ringer-HEPES buffer, before putting the Origami on
it. Still we didn´t see any Origami (data not shown). The interference
factor in the Krebs-Ringer-HEPES buffer could be the lack of magnesium
or the calcium. Therefore we first tested if the Origami are stable in
Origami in Krebs-Ringer-Hepes buffer with 12.5 mM magnesium. Still we
didn´t see any Origami (data not shown).
3. Future prospects
Calcium has the same charge as magnesium, but the ionic radius of
magnesium is much bigger, which could lead to deformed and instable
Origami. Therefore it could be also tested if the Origami are stable in
Krebs-Ringer-HEPES buffer with 12.5 mM magnesium, but without calcium.
Because the buffer has also to be suitable for the cells, the stability
of the Origami in phosphate buffer without calcium should be tested.
 Literature Literature
- Paul W. K. Rothemund: Nature 440, 297-302 (16 March 2006)
|  "
"






