|
Home
The Team
Project Report
Parts
Modeling
Notebook
Safety
CoLABoration
|
_cloning strategy
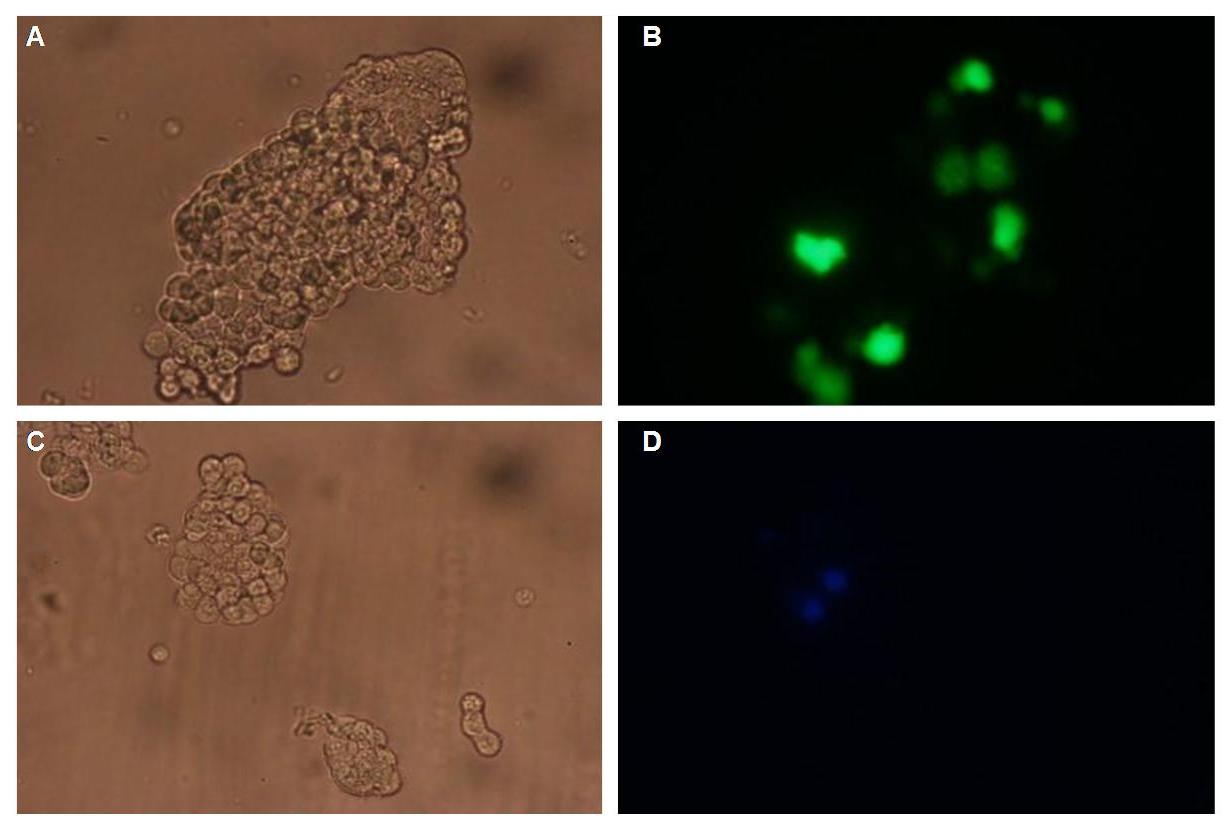
To form different types of synthetic receptor constructs a principle of a building set was used. There were two different forms of an extracellular binding protein and four different components of an intracellular signal transduction reporter protein, Cerulean CFP, Split Venus YFP, β-Lactamase and Luciferase. These reporters were used as Split-proteins to achieve that there is only detection when two receptors come together and form a cluster. With this strategy a signal is only given by two binding-molecules which are next to each other as it is realized in the DNA-origami structure or the NIP and Fluorescein coupled BSA.
The whole receptor construct consists out of a signalpeptide, an extracellular receptor domain, a GGGS-linker followed by transmembrane region and an intracellular split-reporter-protein. The signalpeptide-sequence according to the EGF-Receptor erbb1 ensures the transport of the protein construct to the cytoplasmamembrane. A GGGS-linker is necessary to create a distance between membrane and the recognition-site to overcome cell surface structures as glycoproteins and glyclipids. The sequence of the transmembrane region is appropriate to that of the EGF-Receptor erbb1 as well. The C-terminal split-parts of Venus-YFP and Cerulean CFP were not directly fused to the transmembrane-region. To get a little bit more flexibility in binding to the N-terminal part a Split Fluorophor-linker was assembled in between.
Almost all parts were ordered by genesynthesis in a pMA-vector system which is adapted for cloning BioBrick parts. All possible variations of the receptor constructs were fused together in the pMA-vector and afterwards inserted into a transfectionvector.
The transfectionvector is a BioBrick 2007 part (BBa_J52017 http://partsregistry.org/Part:BBa_J52017 ) and includes a kanamycin and ampicillin resistance cassette. There is a pMB1 Ori and the multiple cloning site consist out of the BioBrick Prefix and Suffix restriction enzymes (Prefix: EcoRI, NotI, XbaI/ Suffix: SpeI, NotI, PstI) followed by an eukaryotic terminator sequence. To achieve a strong expression a constitutive promoter construct was needed. Because of this Cauliflower Mosaik Virus [CMV] promotor was amplified by PCR using the BioBrick part BBa_J52038 (http://partsregistry.org/Part:BBa_J52038) and primers below-mentioned. The forward primer bound in the iGEM-prefix, while reverse primer bound at the end of the CMV-coding region. The reverse primer also contained an overhang to get the iGEM-suffix directly to the end of the promoter sequence which later allowed the cloning into the transfectionvector. CMV promoter was cloned into the vector by using the EcoRI and PstI restriction sites. To test the expression activity of the vector with CMV promoter the gene of the yellow fluorescent protein [YFP] was cloned behind the promoter region by using XbaI, PstI restriction enzymes for the YFP insert and SpeI, PstI for the vector to open Biobrick suffix. The tranfsfection of the resulting plasmid into 293T cells shows fully functionality.
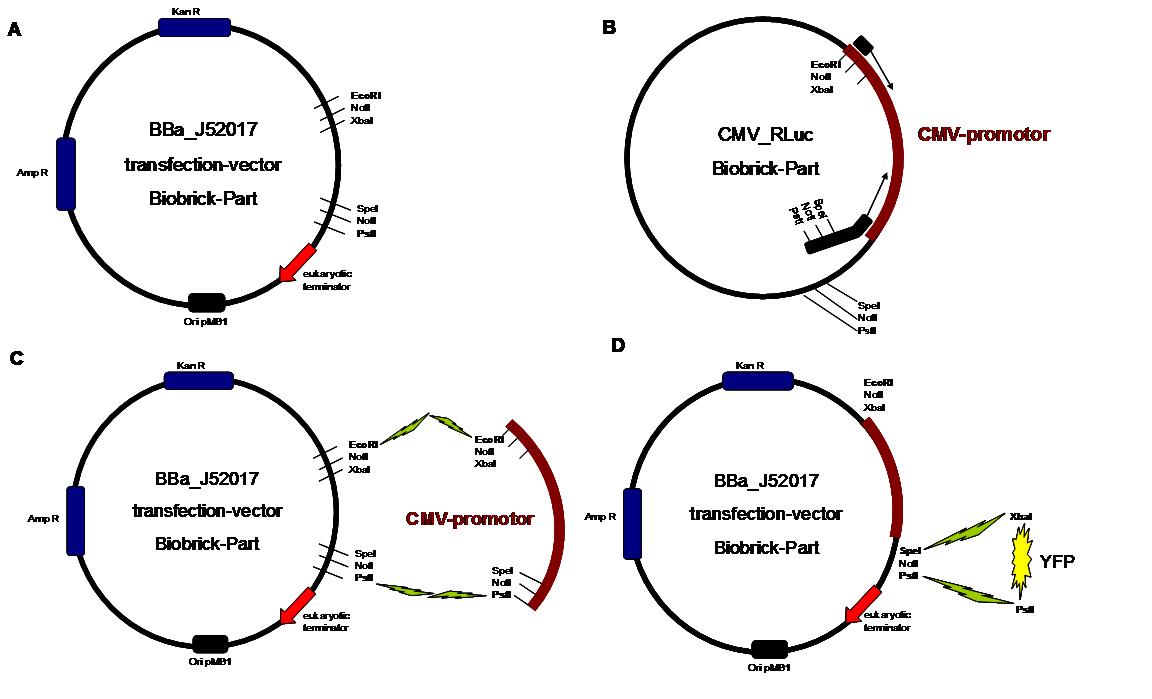
Figure 1 . A shows the Biobrick part BBa_J52017. The transfectionvektor for eukaryotic cell systems has an ampicillin- and a kanamycin resistance cassette. The multiple cloning site contains the Biobrick standard restriction sites EcoRI, NotI, XbaI, SpeI, NotI, PstI followed by an eukaryotic terminator sequence. B The CMV-promotor fragment was obtained by PCR with the Biobrick BBa-J52038 template. C The PCR product was cloned into the transfectionvector by EcoRI and PstI to get a final eukaryotic transfection-system. D To test the efficiency of expression a gene-fragment coding for the yellow fluorescent protein was put into the vector behind CMV-Promotor.
An overview about the cloning is given in table 1
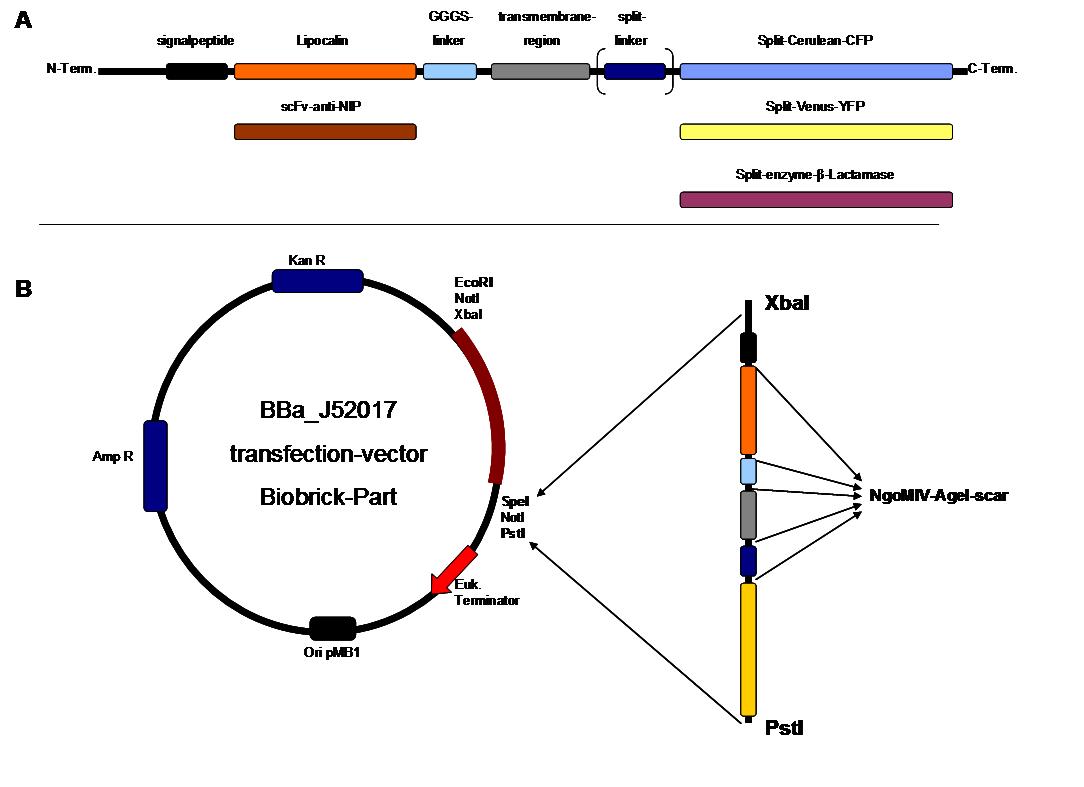
Figure 2 . A figure 2 A gives an overview about the cloning constructs. The N-terminal signal-peptide ensures protein transport to the cytoplasmamembrane. Lipocalin and the scFv-anti-NIP are the extracytoplasmatic parts of the construct to mediate signaltransduction into the cell. The GGGS-Linker keeps a distance to the transmembraneregion to overcome surface structures of the cell and to avoid a total inflexibility. Split fluorophor linker is only necessary for the C-terminal Split parts of Cerulean-CFP and Split-Venus-YFP. The Split enzymes β-Lactamase and Luciferase and the split-fluorophors CFP and YFP are the cytoplasmatic parts of the constructs. If there is a clustering of this synthetic receptor-system caused by the corresponding binding parts of Lipocalin and scFv-anti-NIP the split parts come together to create a functional protein, which allows a detection.
B The different constructions described in figure 2.A were cloned into the transfectionvector system by using the restriction sites XbaI and PstI to ensure a functional ATG-start codon which is part of the XbaI recognition-sequence in the iGEM-prefix
|
Step 1
|
Vector
digestion: EcoRI + PstI
|
Insert
digestion: EcoRI + PstI
|
|
|
BBa-J52017
|
_CMV-promotor
|
|
Step
2
|
Vector
digestion: AgeI+SpeI
|
Insert
digestion: NgoMIV+SpeI
|
|
|
pMA-BBFR
_ SPLIT-Linker
|
C-YFP
|
|
|
C-CFP
|
|
Step
3
|
Vector
digestion: AgeI+SpeI
|
Insert
digestion: NgoMIV+SpeI
|
|
|
pMA-BBFR
_egfR-Tm
|
_ N-β-Lactamase
|
|
|
_ C-β-Lactamase
|
|
|
_ SPLIT-Linker_ C-YFP
|
|
|
_ N-YFP
|
|
|
_ SPLIT-Linker_ C-CFP
|
|
|
_ N-CFP
|
|
|
_ BB058 (Luciferase)
|
|
|
_ BB057 (Luciferase)
|
|
Step
4
|
Vector
digestion: AgeI+SpeI
|
Insert
digestion: NgoMIV+SpeI
|
|
|
pMA-BBFR
_SP
|
_scFv-anti-NIP
|
|
|
_ Lipocalin
|
|
Step
5
|
Vector
digestion: AgeI+SpeI
|
Insert
digestion: NgoMIV+SpeI
|
|
|
pMA-BBFR
_SP_ scFv-anti-NIP
and
pMA-BBFR-+SP_ Lipocalin
|
_GGGS-linker (produced by Klenow fill in)
|
|
Step
6
|
Vector
digestion: AgeI+SpeI
|
Insert
digestion: NgoMIV+SpeI
|
|
|
pMA-BBFR
_SP_ scFv-anti-NIP _ GGGS-Li
and
pMA-BBFR
_ SP_ Lipocalin _
GGGS-Li
|
_
egfR-Tm _ N-β-Lactamase
|
|
|
_
egfR-Tm _ C-β-Lactamase
|
|
|
_
egfR-Tm _ SPLIT-Linker_ C-YFP
|
|
|
_
egfR-Tm _ N-YFP
|
|
|
_
egfR-Tm _ SPLIT-Linker_ C-CFP
|
|
|
_
egfR-Tm _ N-CFP
|
|
|
_
egfR-Tm _ BB058 (Luciferase)
|
|
|
_
egfR-Tm _ BB057 (Luciferase)
|
|
Step
7
|
Vector
digestion:
SpeI + PstI
|
Insert
digestion: XbaI + PstI
|
|
|
BBa-J52017_CMV
|
_SP_ scFv-anti-NIP_GGGS-Li_egfR-Tm_N-β-Lactamase
|
|
|
_ SP_ scFv-anti-NI _GGGS-Li_ egfR-Tm_C-β-Lactamase
|
|
|
_ SP_ scFv-anti-NIP_GGGS-Li_
egfR-Tm_SPLIT-Linker_C-YFP
|
|
|
_ SP_ scFv-anti-NIP_GGGS-Li_ egfR-Tm_N-YFP
|
|
|
_ SP_ scFv-anti-NIP_GGGS-Li_
egfR-Tm_SPLIT-Linker_C-CFP
|
|
|
_ SP_ scFv-anti-NIP_GGGS-Li_ egfR-Tm_N-CFP
|
|
|
_ SP_ scFv-anti-NIP_GGGS-Li _ egfR-Tm_BB058 (Luciferase)
|
|
|
_ SP_ scFv-anti-NIP_GGGS-Li _ egfR-Tm_BB057 (Luciferase)
|
|
|
_ SP_ Lipocalin _GGGS-Li_ egfR-Tm_N-β-Lactamase
|
|
|
_ SP_ Lipocalin _GGGS-Li_ egfR-Tm_C-β-Lactamase
|
|
|
_ SP_ Lipocalin _GGGS-Li_
egfR-Tm_SPLIT-Linker_ C-YFP
|
|
|
_ SP_ Lipocalin _GGGS-Li_
egfR-Tm_N-YFP
|
|
|
_ SP_ Lipocalin _GGGS-Li_
egfR-Tm_SPLIT-Linker_ C-CFP
|
|
|
_ SP_ Lipocalin _GGGS-Li_
egfR-Tm_N-CFP
|
|
|
_ SP_ Lipocalin _GGGS-Li__ egfR-Tm _ BB058 (Luciferase)
|
|
|
_ SP_ Lipocalin _GGGS-Li__ egfR-Tm _ BB057 (Luciferase)
|
METHODS
_________________________________________________________________________________________________________________
The cloning was started with a preparative digestion of the DNA-Plasmids. To clone fusion parts the vector constructs were digested with AgeI and PstI to open the Biobrick suffix. The inserts were digested with NgoMIV and PstI. For cloning into the transfection-vector the enzymes SpeI and PstI were used for vector and XbaI, PstI for insert to keep up the ATG-start codon in the XbaI restriction site of the biobrick suffix. All restriction-enzymes were ordered from New England Biolabs. After digestion the DNA-fragments were separated on a 1% agarose gel. The DNA-band of interest was isolated and purified with the QIAGEN QIAquick Gel Extraction Kit. For the ligation a 3 molar excess of the insert was put together with the vector-fragment and ligated with a Quick ligase (New England Biolabs). After half an our at room temperature the DNA was transformed to chemical competent E.coli strain XL1 cells, plated on 2YT-agar-plates and incubated at 37°C over night. After picking clones and growing in 5ml LB-medium, the plasmid DNA was isolated by QIAGEN QIAprep Spin Miniprep Kit. A test digestion was prepared with about 0,5µg Plasmid DNA and NotI restriction enzyme to isolate the fusion-protein from the vector and to control if the expected bands were obtained. After a positive result the clones were sent to GATC-Biotech for sequencing.
The GGGS-Linker was produced by Klenow -fill-in-PCR. Two primers were designed align to each other at 60°C and filled to a complete dobble-strand by Klenow Polymerase fragment.
Digestion Protocol
- about 2µg Plasmid-Prep in 20µl
- 2 µl NEB Buffer 10x
- 1 µl NEB enzyme 1 (NgoMIV, AgeI, XbaI, EcoRI)
- 1 µl NEB enzyme 2 (PstI, SpeI)
- 0.2µl BSA 100x
Ligation
- 10µl volume of vector and insert DNA (about 50ng vector-DNA)
- 1 µl DNA Quick Ligase (New England Biolabs)
- 10 µl Quick Ligase Buffer
Analytic digestion
- about 0.5 µg Plasmid-DNA in 5µl
- 5µl H2O
- 0.5 µl NotI
- 1µl NEB-Buffer
- 0.1 µl BSA
Transformation
- Competent cells (100µl) werde defrosted on ice
- 10µl of the ligation was added
- DNA and cells werde mixed softly
- Incubation on ice for 20-30 min
- Heat shock at 42°C for 40 sek
- cells were cooled down on ice for 5-10 min
- 900µl sterile 2YT Medium was added
- Incubation at 37°C for 60-70 min (shaker)
- cells werde plated on 2YT-agar-plates with antibiotics
Klenow fill in reaction
- 25pmol forward primer
- 25pmol reverse primer
- 0.5 µl Klenow-fragment without exonuclease activity (Fermentas)
- 2µl Klenow Buffer
- 1µl dNTPs
- Add H2O to a volume of 20µl
program: 94°C for 3min, cool down to 37°C, adition of klenow enzyme, 37°C for 1 hour

 
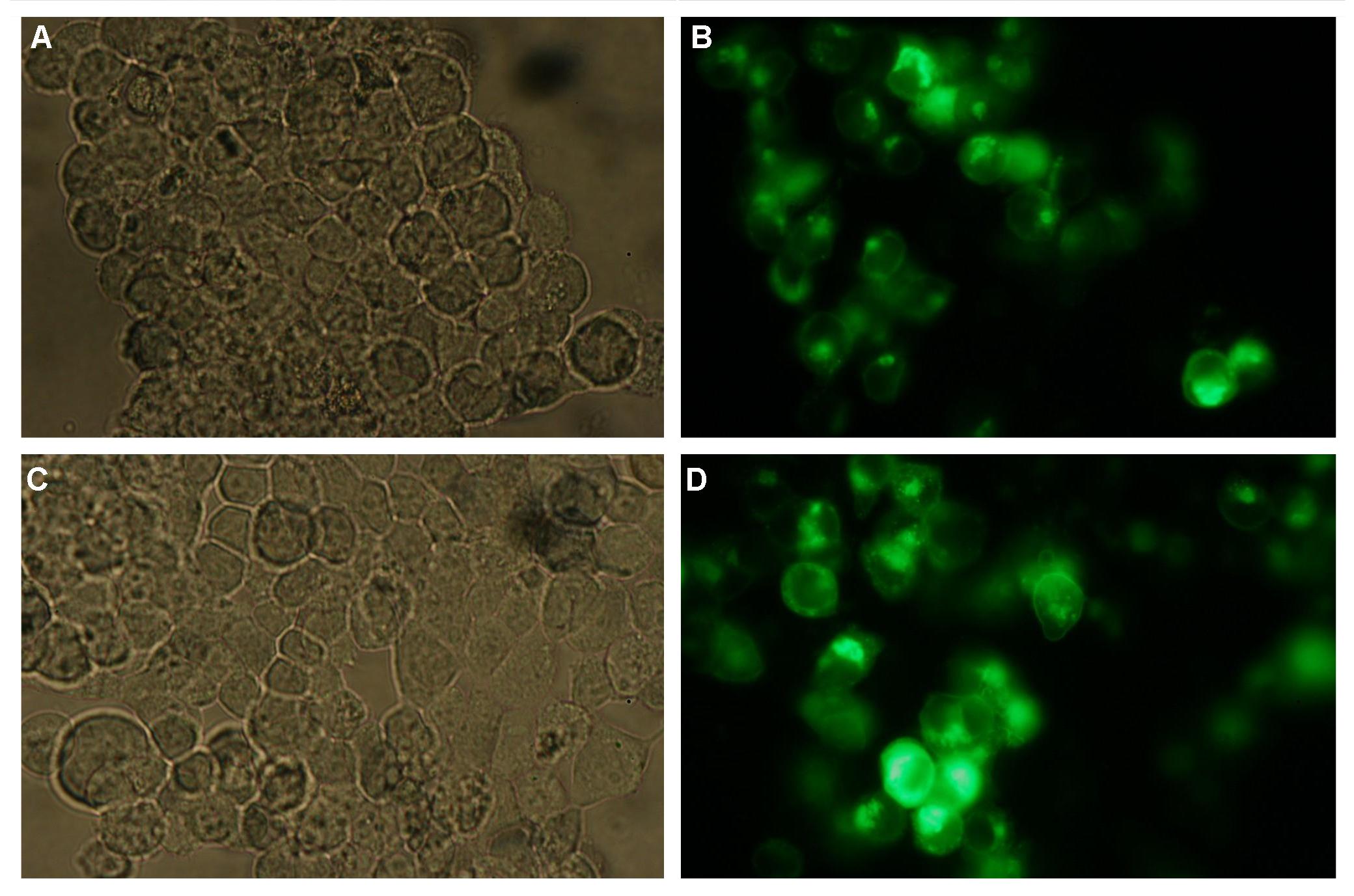
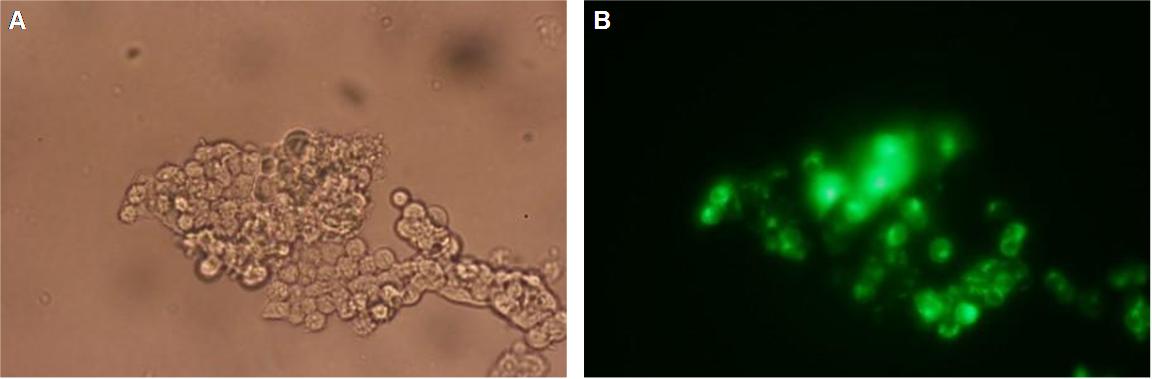
|
 "
"








