|
_july
Jul. 4th 2008
Assessment of Origami-Pool
We mixed all oligos, except for the modified, the left and the right borders and the remainders, together .
1) Oligos modified with fluorophores:
1. board:
r-3t8f
r-5t10e
r-5t12e
2) Oligos modified with NIP:
1. board:
r-7t6f
r-7t8f
r-7t10f
r-5t8f
r-3t10e
r-7t6e
3. board:
r7t22e
3) Left border: 1.board, 1.line
4) Right border: 3.board, 2.line
5) Remainders: 3.board, 3.line
Jul. 6th 2008
Assessment of cell cultures (DYT-Medium; ER 2738-cells)
50ml DYT-Medium
+ 50 TET (25ng/ml -> 1:1000)
+ pick ER 2738-cells (from -80 degree freezer)
-> shake at 37 degree over night
Jul. 7th 2008
1) Thinning down overnight culture to OD~0.1
1:10 -> 0.66 = OD 660
5000µl = 0.66
x = 0.1 -> x = 758µl ~ 760µl in 50ml
We mixed 760µl cell culture with 50ml DYT and shaked both at 37°C
|
t / min |
Assay |
OD (600) |
|
20 |
1 |
0,05 |
|
60
60 |
1
2 |
0,28
0,17 |
|
75
75 |
1
2 |
0,37 *
0,28 |
|
95 |
2 |
0,39 * |
*inoculation of the culture
2) Inoculation of a cell culture with M13mp18 phages
~ 50ml cellculture
+
5µl M13mp18 phages
(from -80°C)
-> 4h shaking at 37°C
3) Phage precipitation
1) centrifuging of the phage-solution at 5000 x g for 20
min.
2) after decanting supernatant dessolve the pellet
in 7ml (1/7 of the supernatant volume) PEG/NaCl
-> The precipitation occurs over night at 4°C
Jul. 8th 2008
1) Isolation of M13 phages
see Methods: Isolation of M13mp18 phages from the cellculture
2) Measurement of the phage titer
-Photometer, Program: Spectrum Measurement
-Absorption at 269nm
-Measurement with Phage-Cuvette!
-Thin down phages 1:30 or 1:40
-Formula to calculate the Phagetiter:
Phagetiter = (A269 * 6 * 10^16 * dilution factor) /(number of nucleotids in phage genom)
Nucleotids in M13mp18-Phage genom = 7249 bp
Solution 1: (0.36*6*10^16*30) / 7249 = 8.9*10^13 phages/ml
Solution 2: (0.33*6*10^16*30) / 7249 = 8.2*10^13 phages/ml
->the unit phages/ml have to be translated in µg DNA/ml because by purifying there should be at most 100µg DNA in a filter (Montage® PCR Centrifugal Filter Devices)
3)Translation in µg DNA/ml
Solution 1: 8.9*10^13 phages/ml ~ 8.9*10^13 particle/ml
8.9*10^13 particle = X1
6.022*10^23 particle = 1Mol
X1 = 1.5*10^-10Mol
1 Mol = 2.2*10^6g DNA
1.5*10^-10Mol = X2
X2 = 3.3*10^-4g DNA -> 330µg DNA/ml
Solution 2:
6.022*10^23 particle = 1Mol
8.2*10^13 particle = X1
X1 = 1.4*10^-10Mol
1 Mol = 2.2*10^6g DNA
1.4*10^-10Mol = X2
X2 = 3.0*10^-4g DNA -> 300µg DNA/ml
-> So we can use 250µl from each solution per round and filter
4) Isolation of the phage DNA with QIAprep M13-Kit
- 1ml phages + 10µl MP-buffer, 2min. rest
- split in 4 filters inside an
Eppendorf-tube each 250µl
- centrifuge 8000g for 15sek.
- add 700µl MLP-buffer and centrifuge at 8000g for
15sek., pour away buffer
- add 700µl MLP-buffer, 1min. rest, centrifuge at
8000g for 15sek., pour away buffer
- add 700µl PE-buffer, centrifuge at 8000g for
15sek., pour away buffer
- for drying, centrifuge again
Elution: put filters in fresh eppendorftubes and add
100µl autoclaved Millipore-water on top of the membrane, inkubate for 10min.
- centrifuge out the water and the solute DNA (8000xg
for 30 sek.)
-> quantify DNA-concentration by drop...:
Solution 1:448,4ng/ml
Solution 2:423,4ng/ml
Jul. 10th 2008
1) Origami(Simone+Michael)
Origami without remainder
7.28µl------Oligos-delta-10(without the 10Oligos which could be marked)
1.18µl------M13pm18 phage DNA
0.4 µl------10 Oligos (Oligos which could be marked-> here we used the unmarked)
1.0 µl------MgAcetat(50x12,5mM Mg2+)
-> add aqua-dest to 50µl
Origami with remainder
7.28µl------Oligos-delta-10(without the 10Oligos which could be marked)
1.18µl------M13pm18 phage DNA
0.4 µl------10 Oligos (Oligos which could be marked-> here we used the unmarked)
0.4 µl------remainders
1.0 µl------MgAcetat(50x12,5mM Mg2+)
-> add aqua-dest to 50µl
The Origami were produced in a eppendorf Master cycler personal. Therefore they were heated up to 95°C for 7 minutes an then slowly cooled down (0,3°C/s) to 20°C.
Setting Master cycler
1. T= 95,0°
0:07:00
2. T= 90,0°
0:01:00
-0,5° +0:00
R=0,3°/s +0,0°/s
3. GOTO 2
Rep 70
4. T= 55,0°
0:01:00
-0,5° +0:00
R=0,3°/s +0,0°/s
5. GOTO 4
Rep 70
6. HOLD
20,0° Enter
7. end
2) Purification of the DNA-origamis with Montage® PCR Centrifugal Filter Devices (Michael+Simone)
To purify the DNA-Origamis of the unbound DNA-oligos we used
Montage® PCR Centrifugal Filter Devices (Millipore).
- The Montage® PCR Centrifugal Filter Devices were
labeled and put in 1,5ml Eppendorf Tubes. The purple side has to be on
top.
- To clean the Filter of remaining Glycerin: 450µl
TAE/MgAcetat (1x filtered) was put on top of the filter and for
15minutes at 1000g (g=rcf) centrifuged.
- The liquid in the Eppi was removed.
- 400µl TEA/MgAcetat + 45µl DNA-origami
were put on top of the filter and again centrifuged for 15minutes at
1000g (g=rcf).
- The liquid in the Eppi was removed.
- To wash of all unbound DNA-oligos: 400µl
TEA/MgAcetat was put on top of the filter and for 15minutes at 1000g
(g=rcf) centrifuged.
- To release the DNA-origamis of the filter put
100µl TAE/MgAcetat on top of the filter and leave it at room
temperature at least for 2 minutes. (not to long, because the filter
shouldn´t run dry)
- The Montage® PCR Centrifugal Filter Devices were
put upside down (the purple side has to be on bottom) in one of the
special Invert Spin tubes form Millipore and for 3 minutes at 1000g
centrifuged.
->The Origami were stored in the fridge(4°C).
Jul. 11th 2008
1) AFM measurement (Michael+Simone+Daniel)
To see if the Origami well formed and an atomic force microscope (AFM) was used.

->all the Origami(with and without NIP) are formed well. So the remainders are not needed!
Jul. 17th 2008
1) DNA-origamis in
different ratios
This measurement was used to test if DNA-origami can be made at
a ratio of DNA-oligos to M13mp18 DNA smaller than 1:20.
Three ratios were tested:
Sample 1: DNA-oligos to M13mp18 DNA -> 1:20 (control)
Sample 2: DNA-oligos to M13mp18 DNA -> 1:10
Sample 3: DNA-oligos to M13mp18 DNA -> 1: 5
We used different amount of M13mp18 DNA per sample!
- 1:20; 4nM M13mp18 DNA
- 1:10; 8nM M13mp18 DNA
- 1:5; 12nM M13mp18 DNA
2) Mastercycler
Samples were putted in the Mastercycler as before.
Program -> Origami 2
3) purification of the
DNA-origamis
To remove the unbound DNA-oligos the samples were washed as before
-> protocol 07-10-2008
Changes:
To prove whether all of the DNA-origami were washed off the Millipore
we repeated step 5 and 6 of the protocol.
4) Measurement on the AFM

Jul. 22nd 2008
1) Test Ringer-Solution
With this essay, the stability of the DNA-Origami in
Ringer-Solution was tested.
4 probations were taken:
1. Probation:
we took M13mp18 DNA and DNA-oligos in a ratio of 1:20 (protocol from
07-17-2008) and washed the origami in the Millipore-filter with
TEA/MgAcetat (control of 1:20)
2. Probation:
we took M13mp18 DNA and DNA-oligos in a ratio of 1:20 (protocol from
07-17-2008) and washed the origami in the Millipore-filter with the
Ringer-Solution (1:20 with Ringer)
3. Probation:
we took M13mp18 DNA and DNA-oligos in a ratio of 1:5 (protocol from
07-17-2008) and washed the origami in the Millipore-filter with
TEA/MgAcetat (control of 1:5)
4. Probation:
we took M13mp18 DNA and DNA-oligos in a ratio of 1:5 (protocol from
07-17-2008) and washed the origami in the Millipore-filter with the
Ringer-Solution (1:5 with Ringer)
The origami were made as before -> protocol from 07-17-2008
The origamis were purified as before. Except for the probations we
wanted to test the Ringer-Solution we used the Ringer-Solution instead
of TEA/MgAcetat -> protocol from 07-10-2008
If the origamis are also stable in the Ringer-Solution we should see
well formed DNA-origami in the AFM.
Jul. 23rd 2008
1) Measurement on the AFM
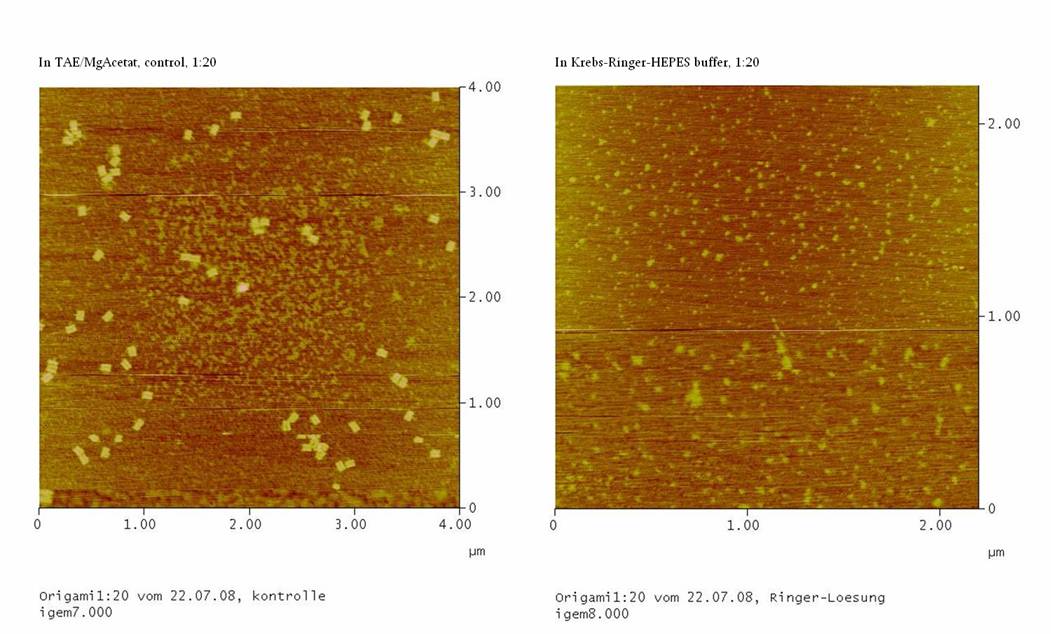
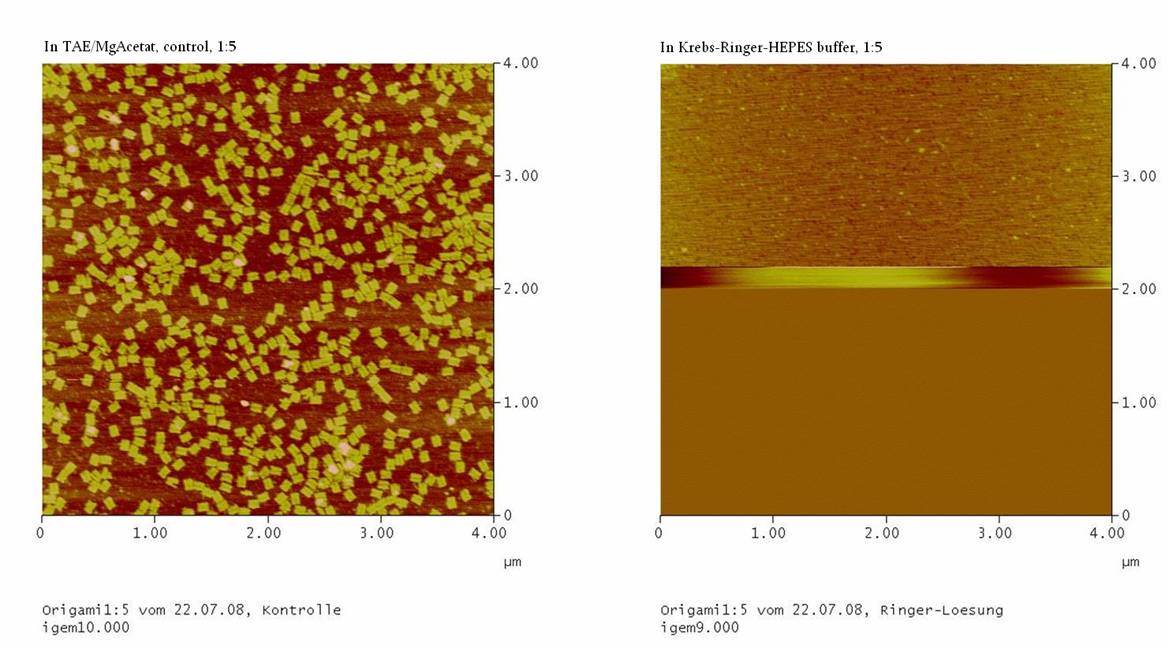
We couldn’t see any DNA-origami!!!
-> may be the origami are not stable in the Ringer-Solution
or
-> The NaCl in the Ringer-Solution disturbed the binding between
the glimmer-surface and the origami. To test this, the concentrations
of the four probations were measured
……… in the Nano-drop. If the
concentrations of the probations with Ringer-Solution is comparable to
the concentrations of the controls, than we should purify the
DNA-origami again, using TAE/MgAcetat as buffer.
2) Measurement on the
Nano-Trop
| Sample |
ng/µl |
A260 |
A280 |
260/280 |
| 1:20 with Ringer |
4.19 |
0.105 |
0.075 |
1.40 |
| 1:20 with Ringer |
4.46 |
0.111 |
0.078 |
1.43 |
| 1:5 with Ringer |
18.33 |
0.458 |
0.277 |
1.65 |
| 1:5 with Ringer |
15.96 |
0.399 |
0.248 |
1.61 |
| control of 1:20 |
5.94 |
0.148 |
0.097 |
1.53 |
| control of 1:5 |
14.16 |
0.354 |
0.200 |
1.77 |
Probation 2 showed nearly the same concentration as probation 1.
Probation 4 showed nearly the same concentration as probation 3.
-> There might by well formed origami in the Ringer-Solution. To
test this further, the probations with Ringer-Solution were purified
and buffered with TAE/MgAcetat.
3) buffer the DNA-origami
from Ringer-Solution in TAE/MgAcetat
The DNA-origamis in Ringer-Solution were purified as before (->
purification protocol 07-10-2008)
Changes of the protocol:
-> Instead of 45µl of Origami we putted
100µl of our DNA-origami in Ringer-Solution (07-22-2008,
probation 2 and 3) on the Filter (Step4).
4) Order new
Montage® PCR Centrifugal Filter Devices
We ordered new Montage® PCR Centrifugal Filter Devices at
Millipore online.
Jul. 24th 2008
1) DNA-Origamis with NIP
and fluorophor
For the measurement at the fluorescence microscope we prepared some
DNA-oligos with NIPs and fluorophor.
First we wanted to test if the DNA-origami with NIPs bind to the cells.
For this we prepared DNA-origami coupled with NIPs and fluorophor and
DNA-origami coupled with fluorophor but without NIPs (negative
control). Ratio M13mp18 DNA to Oligos 1:5.
As fluorophor we took Alexa488 (λexc= 488 nm,
λem= 515 nm) and we used all of the 7NIPs.
Second we wanted to test if the DNA-Origami with NIPs induces cellular
signalling by measuring the calcium influx. For this we prepared
DNA-origami with NIPs and DNA-origami without NIPs (negative control).
Ratio M13mp18 DNA to Oligos 1:5.
Jul. 29th 2008
1) Measurement at the AFM
We measured the origami which we produced in Ringer-Solution. Because
we didn´t see any origami in the last measurement (07-23-08)
we buffered them in TAE/MgAc and measured them again.
BILD
We couldn’t see any origami only some greasy spots which were
smaller (50-70 nm) then the right ones (100 nm).
Improvements:
1.) We could put more Mg into the Ringer-Solution to boost the
DNA-backbone.
2.) If the T-cells tolerate the TAE/MgAc in small concentrations we
could use this buffer to produce the origami.
2) Purification of the DNA-origamis
We purified the origami with fluorophor, with fluorophor and NIPs, just
with NIPs and without NIPs or fluorophor (Control) from 07-24-2008.
-> Protocol from 07-10-2008
Changes: instead of 45 µl we used 65 µl of the
DNA-origami.
Jul. 31st 2008
T-cell staining
1) The T-cells of 2 falcontubes were spun down at 1200rpm for 5 min. The supernatant was thrown away.
2) The pellets were resuspended in 25ml RPMI and again spun down at 1200rpm for 5 min. The supernatant was thrown away.
3) The pellet was resuspendet in 11ml RPMI (with 1% FCS). For two new cultures we put in both petri dish 5ml of the cell suspension.
The remaining 1ml was spun down (as before). The supernatant was thrown away.
4) 1ml RPMI with 1%FCS, 0,5µl Fura2AM and 0,625µl Pluronic were put in an 1,5ml Eppendorf-Tube and mixed on the vortexer.
With this mixture we resuspended the pellet in the falcon tube(cells).
-> shaking for 30minutes at room temperature.
2) Wash the cells and buffer it in the
Ringer-Solution
For the measurement we have to buffer the cells in Ringer-Solution, because
the phenol red in the RPMI could disturb the measurement. Therefore we washed
the cells with 10ml of Ringer-Solution.
10ml Ringer-Solution was put in the falcon tube with the cells and
centrifuged at 1200rpm for 4minutes. (Normally the cells should be centrifuged
for 5minutes at 1200rpm.). The supernatant was thrown away and the pellet was
resuspended in 100µl Ringer-Solution. 10µl of the cells were put in each well of
the slide.
Changes: Normally the cells should be washed twice, but by throwing away the
supernatant we lost lots of cells, so we didn´t wash it a second time. Also the
cell pellet should be resuspended in 200µl instead of 100µl.
3) Calcium measurement on the
microscope
Activators:
1) 1µl NIP15 – BSA in 100µl Ringer-Solution.
2) 1µl NaVO3 in 100µl Ringer-Solution. NaVO3 inhibits
the Phosphotase, which dephosphorylate some cell structures this leads to
calcium influx (positive control).
3) 1µl Anti-CD28-antibody in 100µl Ringer-Solution.
Measurement:
The calcium influx was measured at 340nm and 380nm. The quotient of both
(340/380) changes when the cells absorb the calcium
from the outside. The calcium is absorbed when the TCR gets activated.
When Fura2 is bound to calcium it shows more signal at 380nm and less signal
at 340nm.
1. + 1µl NIP15 – BSA in one well -> nothing happened
2. +10µl NIP15 – BSA in one well -> nothing happened
3. +10µl Pervanadat in one well -> nothing happened
-> We guess that the cells were not labelled properly, because normally
the FURA 2 stained cells bleach over time. But our cells didn´t bleach at
all.
Because of this we stained the cells again. We put 10µl of Fura2AM on top of
the unused cells in the wells and incubated it for 30minutes at room
temperature. We didn´t wash the cells but put directly the activators on it.
Measurement:
1. 10µl Pervanadat -> we could see a change of
fluorescence which was most likely caused of the calcium influx, because the activity at 380nm increased while the signal at 340nm decreased.
4) Measure the binding of DNA-Origami to the cells
We wanted to test if the DNA-origamis with 7NIPs and flourophor and the
DNA-origami without NIPs but with fluorophor (negative control) from 07-24-2008
bind to the T-cells.
The cells were measured on an inverse fluorescent microscope (excitation:
488nm, measured at 500-550nm).
We also used the FURA stained cells because we didn´t have any others, but
better is to use unstained cells.
Test solutions (in 1,5ml eppendorf tube):
1. 10µl of stained cells (-> 1) T-cell staining)
+ 10µl of the DNA-origamis with NIPs and flourophor(Alexa
488)
2. 10µl of stained cells (-> 1) T-cell staining)
+ 10µl of the DNA-origamis with flourophor (Alexa 488), but without
NIPs
-> Both solutions were mix in a 1,5ml eppendorf tube and then out on the
wells.
Procedure:
To see if the cells are fine we put 10µl of the stained cells directly in one
well. At the same time we put the test-solution 1 on another well. We measured
the cells. When we looked at these two probations we saw that not the whole well
was covered with the solutions so we decided to put more volume in it. Therefore
we put directly 10µl Ringer-Solution in the well with the stained cells only and
10µl of the DNA-Origamis with NIPs and fluorophor in the corresponding well.
Also we put 10µl of DNA-origamis with flourophor, but without NIPs in the
corresponding eppendorf tube, mixed it and also put it on a well.
Outcomes:
In both probations (DNA-origamis with and without NIPS) bound to the T-cells
surface. In the beginning it seemed that the DNA-Origamis without NIP do not
show as much signal as the one with NIPs, but this could basically be caused of
to thinks:
1. They weren´t illuminated as long as the T-cells with NIP coupled
DNA-Origami.
2. The Cells with the DNA-origamis with flourophor, but without NIPs were
much better mixed than the other ones, because we could mix the whole test
solution before putting it on the well.
improvements in general:
1. We should always bring some unstained cells, to have some comparison. For
example to see if the stained cells are probably stained or if the fluorescence
we see comes from the cells autofluorescens
2. We should use 20µl of the cell-culture (or 10µl of cells with 10µl of
Ringer-Solution) so that the whole well is covered with liquid.
|  "
"




