 "
"
Team:ETH Zurich/Project/Medal Relevant
From 2008.igem.org
m (→Modeling Framework) |
m |
||
| Line 16: | Line 16: | ||
=== Gold Relevant === | === Gold Relevant === | ||
| + | |||
| + | ==== Analysis, modeling, and simulation of BioBrick Parts or Devices ==== | ||
We propose a novel method of random gene deletion and chemostat-based selection of species with a reduced genome. For this we provide an algorithm described below. | We propose a novel method of random gene deletion and chemostat-based selection of species with a reduced genome. For this we provide an algorithm described below. | ||
[[Image:Gold_Algorithm.jpg|frame|none|Genome Reduction Algorithm]] | [[Image:Gold_Algorithm.jpg|frame|none|Genome Reduction Algorithm]] | ||
| - | ==== Modeling Framework ==== | + | ===== Modeling Framework ===== |
This algorithm requires a modeling framework consisting of four main parts: | This algorithm requires a modeling framework consisting of four main parts: | ||
# [[Team:ETH_Zurich/Modeling/Genome_Static_Analysis|Statistical Analysis]] of DNA fragments produced by Restriction Enzymes | # [[Team:ETH_Zurich/Modeling/Genome_Static_Analysis|Statistical Analysis]] of DNA fragments produced by Restriction Enzymes | ||
| Line 26: | Line 28: | ||
# [[Team:ETH_Zurich/Modeling/Switch_Circuit|Switch generator]] for short-time Restriction Enzyme Expression | # [[Team:ETH_Zurich/Modeling/Switch_Circuit|Switch generator]] for short-time Restriction Enzyme Expression | ||
| - | ==== Detailed description of the Modeling Framework ==== | + | ===== Detailed description of the Modeling Framework ===== |
# '''Restriction Enzyme Analysis''' | # '''Restriction Enzyme Analysis''' | ||
#:We want to identify a restriction enzyme for genome reduction that maximizes the probability of genome reduction and minimizes the probability to hit an (known) essential gene. Therefore, given a genome data of an E. Coli strain, the genome is digested using ca. 700 different restriction enzymes (with different recognition patterns). The resulting fragments are analyzed using the available annotation of the genome. The number of genes disrupted is calculated for each fragment. Statistical measures of the restriction enzyme effects, such as average size of the fragments and its variance, and the average number of (all and only essential) genes and its variance are calculated. According to these results, the most suitable restriction enzyme is chosen. | #:We want to identify a restriction enzyme for genome reduction that maximizes the probability of genome reduction and minimizes the probability to hit an (known) essential gene. Therefore, given a genome data of an E. Coli strain, the genome is digested using ca. 700 different restriction enzymes (with different recognition patterns). The resulting fragments are analyzed using the available annotation of the genome. The number of genes disrupted is calculated for each fragment. Statistical measures of the restriction enzyme effects, such as average size of the fragments and its variance, and the average number of (all and only essential) genes and its variance are calculated. According to these results, the most suitable restriction enzyme is chosen. | ||
| Line 36: | Line 38: | ||
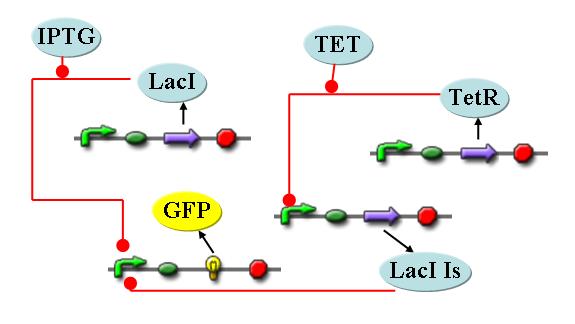
#:Using a novel pulsing mechanism consisting of two signals, start signal, which initiates the expression of restriction enzymes and stop signal, which switches off the expression, we are able to delete genome fragments in vivo. To simulate this system we developed a switch curcuit which works as follows (see [[Media:SimpleCircuit.jpg| Switching Curcuit]]) by inducing the system with IPTG, the restriction enzyme (RE), which is under control of lacI, can be expressed. In order to stop the expression the system is induced with tet, which inhibits the binding of tetR to LacIS (a mutant of LacI, which is not inducable by IPTG), therefore activates the LacIS expression and consecutive termination of RE expression. This curcuit is modeled by ca. 40 reactions and is simulated using ODE solver and stochastic simulations. | #:Using a novel pulsing mechanism consisting of two signals, start signal, which initiates the expression of restriction enzymes and stop signal, which switches off the expression, we are able to delete genome fragments in vivo. To simulate this system we developed a switch curcuit which works as follows (see [[Media:SimpleCircuit.jpg| Switching Curcuit]]) by inducing the system with IPTG, the restriction enzyme (RE), which is under control of lacI, can be expressed. In order to stop the expression the system is induced with tet, which inhibits the binding of tetR to LacIS (a mutant of LacI, which is not inducable by IPTG), therefore activates the LacIS expression and consecutive termination of RE expression. This curcuit is modeled by ca. 40 reactions and is simulated using ODE solver and stochastic simulations. | ||
| - | ==== Summary of the Algorithm and Interplay of Frameworks Componets ==== | + | ===== Summary of the Algorithm and Interplay of Frameworks Componets ===== |
First tree steps initialize and prepare the system for gene deletions and growth simulations. | First tree steps initialize and prepare the system for gene deletions and growth simulations. | ||
Revision as of 17:53, 27 October 2008
Medal Relevant IssuesSilver RelevantGold RelevantAnalysis, modeling, and simulation of BioBrick Parts or DevicesWe propose a novel method of random gene deletion and chemostat-based selection of species with a reduced genome. For this we provide an algorithm described below.  Genome Reduction Algorithm Modeling FrameworkThis algorithm requires a modeling framework consisting of four main parts:
Detailed description of the Modeling Framework
Summary of the Algorithm and Interplay of Frameworks ComponetsFirst tree steps initialize and prepare the system for gene deletions and growth simulations. In the first step the genome data are analyzed in order to find a most suitable restriction enzyme for random fragments deletion using the “Restriction Enzyme Analysis”- procedure. For the second step the state-of-the art model is adjusted by introducing a selective pressure due to the genome size. Thirdly, initial population consists of one type, namely wildtype. The next steps of the algorithm perform in depth genome fragments deletion and growth simulation. First, we simulate the restriction enzyme expression and the consecutive population decline and occurrence of new mutants with reduced genome, which growth rates can be predicted using Flux Balance Analysis. For these simulations we used the framework parts: “Switch generator” and “Flux Balance Analysis on a Genome Scale Model “. Secondly, the growth simulations are performed using a chemostat model and the distribution of different mutant types after the growth type are obtained. This simulation continues until no better mutants can be generated. Eventually, the genome data of the fastest growing reduced genome mutant can be returned.
|
{kind=link}